Attenuating ETEC Virulence Using a Heat-labile Enterotoxin–blocking Binding Protein

Research Summary
Bacterial enteric pathogens are major contributors to the global burden of diarrheal diseases and the associated consequences for human health including malnutrition, growth stunting, morbidity, and mortality. While mortality from diarrhea has decreased, incidence remains high, and better interventions for preventing disease are needed. Single-domain antibodies (i.e., $\text{V}_\text{H}\text{Hs}$), functioning as target-binding proteins in the gastrointestinal tract, have been proposed as a potential approach to mitigate bacterial pathogenesis. Here, we describe a mitigation strategy where precision binding of a bivalent $\text{V}_\text{H}\text{H}$ to the receptor-binding B-pentamer of heat-labile enterotoxin aggregates the $\text{AB}_5$ toxin and impairs enterotoxigenic Escherichia coli colonization in a flow chamber model simulating the human intestine. The $\text{V}_\text{H}\text{H}$ construct also binds to the structurally similar cholera toxin and effectively abrogates its intestinal cell cytotoxicity in vitro. Based on these results, we believe that targeting virulence could emerge as a new strategy for the management of bacterial enteric pathogens, supporting gut health in at-risk populations alongside vaccination campaigns or in populations without access to vaccines.
Introduction
Diarrheal diseases kill more than one million people globally each year. They disproportionately affect low- and middle-income countries (LMICs) and are the second leading cause of death for children under five years of age.1,2 A major contributor to acute diarrhea in resource-limited settings is the food- and water-borne pathogenic bacterium enterotoxigenic Escherichia coli (ETEC).3,4 ETEC strains exhibit remarkable heterogeneity in their virulence factor repertoire, characterized by diverse combinations of heat-labile enterotoxin (LT), heat-stable toxins (STa and STb), and a wide array of colonization factors (CFAs).5 Further, E. coli exhibits a remarkable capacity for horizontal gene transfer, enabling a continuously evolving population of pathotypes.6,7 This diversity poses significant challenges in identifying key targets for the development of effective control strategies, such as vaccines, with protective effect and broad coverage.8 However, while ETEC colonization and infection are orchestrated by a sequential deployment of these virulence factors, effective delivery of toxins to epithelial cells remains essential to ETEC virulence.9,10
The pathogenic pathway of ETEC is similar to that of Vibrio cholerae, primarily due to their ability to produce two genetically and functionally related enterotoxins: LT from ETEC and cholera toxin (CTX) from V. cholerae.10-12 Phylogenetic analysis suggests that LT and CTX evolved from a common ancestor.11 This is reflected in their shared structural features as $\text{AB}_5$-type toxins, the high sequence similarity between the receptor-binding B-subunits of LT (LTB) and CTX (CTXB) (83% sequence identity), and their common intestinal cell surface receptor, GM1, to which both toxins bind with strong (nanomolar) affinity.13-16 Both LTB and CTXB compromise intestinal barrier function, which enables toxin endocytosis and the intracellular release of the toxic A-subunit.10,17,18 The A-subunits of LT (LTA) and CTX (CTXA) then spur the activation of adenylate cyclase (via $\text{Gs}_{\alpha}$) and facilitate the opening of ion channels, which leads to ion efflux across the intestinal epithelium and ultimately secretory diarrhea driven by osmosis.10,19
Maternal antibodies (IgA and IgG) against LT and CTX found in breast milk confer protection against disease from both ETEC and V. cholerae.18,20 Similarly, we recently demonstrated that camelid-derived single-domain antibodies (i.e., $\text{V}_\text{H}\text{Hs}$) can attenuate V. cholerae virulence by blocking CTX–GM1 binding.21 $\text{V}_\text{H}\text{Hs}$ retain the antigen affinity of full-length antibodies but demonstrate the high pH and protease resistance needed to effectively neutralize intestinal pathogens and their virulence factors in the gastrointestinal tract.22-24 Additionally, their small size and robust format enable efficient, low-cost production using industrially relevant microbial expression systems and biomanufacturing processes.25
In line with the above, a previous study has indicated that oral supplementation of the LT-neutralizing bivalent $\text{V}_\text{H}\text{H}$ BL2.2 can support a healthy and diverse gut microbiota of piglets exposed to ETEC, potentially mitigating post-weaning diarrhea.26 Here, we characterize BL2.2 for its ability to effectively abrogate LT-induced cytotoxicity in human intestinal cells. We characterize the interaction interface between the bivalent $\text{V}_\text{H}\text{H}$ construct and LT and the highly similar CTX, respectively, and detail how natural GM1 receptor binding of both toxins is efficiently blocked by the bivalent $\text{V}_\text{H}\text{H}$ construct. Lastly, we show that this highly efficient blocking of LT significantly reduces the ability of human pathogenic ETEC strain H10407 ($\text{LT}^+$ $\text{ST}^+$) to colonize human Caco-2 cell layers in a flow chamber infection model and preserves epithelial integrity. Our findings thus indicate that neutralizing a single key virulence factor can effectively reduce bacterial pathogenesis, and that toxin-neutralizing $\text{V}_\text{H}\text{H}$ constructs could find utility as tools for limiting the global burden of diarrheal diseases.
Materials and methods
Commercially available materials and reagents
CTX (C8052), CTXB (C9903), and GM1 (G7641) were purchased from Sigma-Aldrich. Lyophilized CTX was dissolved in $\text{dH}_2\text{O}$ to a concentration of $0.5\,\text{mg}\,\text{ml}^{-1}$. CTXB and GM1 were dissolved in phosphate-buffered saline (PBS, pH 7.4) to a concentration of $1\,\text{mg}\,\text{ml}^{-1}$.
Toxin expression and purification
For the molar ratios presented in this study, LTB and CTXB denote a single B-subunit (11.6 kDa) in LTB or CTXB pentamers (58 kDa). The holotoxins LT/CTX (~85 kDa) include the catalytic subunit, LTA/CTXA. LT was expressed and purified as previously described.27 ETEC strain H10407 ($\text{LT}^+$ $\text{ST}^+$) was incubated in CAYE media (Merck, 100060) at 37°C while shaking (200 rpm) for 18 hours. Both cell-bound LT and LT from the cell supernatant were extracted. The supernatant was separated by centrifugation at $4{,}000 \times g$ for 10 min. The remaining cell pellet was dissolved in PBS and sonicated for 7 min using a Fisherbrand™ Model 120 Sonic Dismembrator (Fisher Scientific) to retrieve cell-bound LT. The sonicated cells were centrifuged at $15{,}000 \times g$ for 20 min and the supernatant collected. Cell-bound LT and LT from the cell supernatant were pooled and purified using D-galactose resin (Thermo Scientific, 20372) as previously described.28
LTB-6HIS-3FLAG was produced by E. coli BL21 (DE3) using autoinduction medium (Formedium, AIMTB0205) supplemented with 50 $\mathrm{\mu g}\,\text{ml}^{-1}$ kanamycin at 30°C, while shaking at 150 rpm for 20 hours. LTB was extracted from the periplasm and purified using immobilized metal affinity chromatography (IMAC) as previously explained. Purified LTB was dialyzed against PBS overnight before protein purity was assessed by SDS-PAGE.
For in vitro evaluation of BL2.1 and BL2.2 binding and toxin neutralization capacity, commercially available CTX (Sigma-Aldrich, C8052) and CTXB (Sigma-Aldrich, C9903) were used. For size-exclusion chromatography (SEC) analysis of BL2.1/BL2.2 and toxin interaction analysis, LT and CTX were produced in Vibrio natriegens as previously described.29 Briefly, cells harboring the LT or CTX plasmid were grown at 30 °C in LB-v2 salts medium (LB media supplemented with 204 mM NaCl, 4.2 mM KCl and 23.1 mM $\text{MgCl}_2$) with chloramphenicol (25 $\mathrm{\mu g}\,\text{ml}^{-1}$). When cells reached an $\text{OD}_{600}$ of ~0.8, L-arabinose was added to a final concentration of 0.2% to induce LT/CTX production. After 20 h, proteins were harvested from the culture media by two rounds of centrifugation at $8{,}500 \times g$ for 30 min at 4 °C. The supernatant was filtered, loaded on a D-galactose column (Pierce, Thermo Scientific, 20372), and the protein was eluted using 50 mM Na-phosphate pH 7.4, 200 mM NaCl, 500 mM D-galactose. The LT– or CTX-containing fractions were pooled, concentrated and further purified by SEC using a Superdex 200 increase 10/30 GL column (Cytiva) pre-equilibrated with PBS.
Toxin biotinylation
CTXB and LTB were mixed with No-Weigh NHS-PEG$_4$-Biotin (Thermo Scientific, A39259) at a toxin:biotinylation reagent ratio of 1:2 and incubated at room temperature for 30 min. Excess reagent was reduced at least 125-fold by adding the mixture to 3 kDa molecular weight cut-off (MWCO) protein concentrators (Thermo Scientific, 16311964) together with PBS, followed by centrifugation at $4{,}300 \times g$.
Plasmid construction and $\text{V}_\text{H}\text{H}$ dimerization
The $\text{V}_\text{H}\text{H}$ BL2.1 was originally discovered by Harmsen et al. (2009) and optimized for E. coli expression using the pSANG10-3F vector as previously described.23,30,31 The bivalent $\text{V}_\text{H}\text{H}$ construct BL2.2 was created by genetic fusion of two BL2.1 using a $\text{(GGGGS)}_3$ linker as described elsewhere.26
Expression and purification of $\text{V}_\text{H}\text{H}$ constructs
BL2.1 was produced by E. coli BL21 (DE3) using autoinduction medium (Formedium, AIMTB0205) supplemented with 50 $\mathrm{\mu g}\,\text{ml}^{-1}$ kanamycin at 30 °C, while shaking at 150 rpm for 20 hours. Cells were pelleted by centrifugation at $6{,}000 \times g$ for 10 min and resuspended in periplasmic buffer (25% sucrose with 0.1% lysozyme in PBS). After 30 min incubation at room temperature, an equal amount of cold water was added to the solution and incubated on ice for 10 min. The periplasmic fraction was extracted by centrifugation at $20{,}000 \times g$ for 30 min and purified using IMAC as previously described.21 The eluate was diluted 20 × in 25 mM Na–acetate (adjusted with HCl to a pH of 3.9) and further purified by cation exchange chromatography using a HiTrap SP FF 5 ml column connected to an NGC purification system (Bio-Rad).
BL2.2 was produced at Novonesis laboratories (Bagsværd, Denmark) using microbial fermentation with secretory expression of N-glycosylated protein. The supernatant containing BL2.2 was sterile-filtered and frozen before delivery.
BL3.2 was produced in Komagataella phaffii and purified using IMAC as previously described.21 In brief, an overnight culture (BMGY media) was used to inoculate BMMY media ($\text{OD}_{600} = 1$), which was incubated for 2 days at 30 °C and 160 rpm. Each day, the culture was supplemented with 1% methanol. Prior to purification, the media was centrifuged ($15{,}000 \times g$, 15 min), and the supernatant collected. IMAC purification of the supernatant was carried out using HisPur Ni-NTA resin and IMAC wash buffer at a 1:1 ratio (w:w), eluted using 20 ml IMAC elution buffer followed by 15 ml 1 M imidazole.
Protein concentration was determined using either a Qubit 4 fluorometer (Fisher Scientific, Q33238) and a broad range protein quantification kit (Fisher Scientific, A51292) or a NanoDrop One spectrophotometer (Fisher Scientific, ND-ONE-W). Protein purity was accounted for using visual assessment of SDS-PAGE analysis.
Affinity determination using BLI and SPR
The kinetic parameters of E. coli-expressed, non-N-glycosylated BL2.1 were measured using both bio-layer interferometry (BLI) and surface plasmon resonance (SPR). For BLI experiments, an Octet RED96 instrument from ForteBio was used and data was analyzed as previously described.21 In short, biotinylated BL2.1 (76 nM) was captured by a streptavidin biosensor (Sartorius, 18-5020). The biosensor coated with BL2.1 was then exposed to serial dilutions of four different concentrations (120–0.470 nM) of either CTXB or LTB. Kinetic parameters were determined using the Octet Data Analysis Studio software v12.2.2.26 (ForteBio).
A Biacore 8K+ GoldSeal (GE Healthcare Biosciences) was used for all SPR analysis. The kinetic interactions between BL2.1/BL2.2 and CTXB/LTB pentamers were analyzed both with BL2.1/BL2.2 as the ligand and CTXB/LTB as the analyte, and vice versa. Ligands were immobilized on a CM5 sensor chip (Cytiva, 29149603). All analytes were diluted in HBS-EP+ buffer (Cytiva, BR100669) and evaluated at a wide range of concentrations (1–300 nM) using single-cycle kinetics. Each analysis contained a startup phase of 120 s contact time and 60 s dissociation time with a flow rate of 30 $\mathrm{\mu} \text{l}\,\text{min}^{-1}$ at 25 °C. CTXB was analyzed using a contact time of 180 s and a dissociation time of 1000 s. LTB was analyzed using a contact time of 180 s 120 s and a dissociation time of 10000 s. BL2.1 was analyzed using a contact time of 30 s and a dissociation time of 7000 s. All analytes were evaluated at a flow rate of 30 $\mathrm{\mu} \text{l}\,\text{min}^{-1}$ at 25 °C. Data were analyzed by first subtracting the reference flow cell and HBS–EP+ buffer signals and fitting the data (excluding refractive index and baseline drift) with a global model (1:1 of binding sites). Kinetic parameters were determined using the Biacore Insight Evaluation Software v5.0.18.22102 (Cytiva).
Determination of blocking capacity of toxin-receptor interaction
The ability of BL2.1 and BL2.2 to hinder the interaction between CTXB/LTB pentamers and the ganglioside GM1 receptor was determined using GM1 immobilized onto microplates in a previously described dissociation-enhanced lanthanide fluorescence immunoassay (DELFIA).24,30,32
60 μl of GM1 (Sigma-Aldrich, G7641) was directly immobilized (5 $\mathrm{\mu}\text{g}\,\text{ml}^{-1}$) onto 96-well Immuno Plates (Thermo Scientific, 437111) by incubation overnight. The plates were washed three times with PBS and blocked with PBS supplemented with 3% non-fat milk (M-PBS). BL2.1 and BL2.2 (36 nM) were mixed with biotinylated CTXB or LTB (36 nM) in M-PBS and incubated at 37 °C for 30 min before being added to the GM1-immobilized plate for 1 hour at room temperature. A biotinylated toxin-only (CTXB pentamer) control (36 nM) was included. The plates were washed again, three times with PBS-Tween (0.2%) and three times with PBS, and 100 ng ml−1 of streptavidin-conjugated europium (PerkinElmer, 1244-360) diluted in DELFIA assay buffer (PerkinElmer, 4002-0010) was added. Following 30 min incubation at room temperature, the previous washing procedure was repeated and europium fluorescence activated by adding DELFIA enhancement solution 20 (PerkinElmer, 4001-0010). Intensity measurements (excitation at 320 nm and emission at 615 nm) were carried out using a microplate reader (Victor Nivo) and the blocking capacity determined by relative comparison to the negative toxin-only control.
Competitive binding assay
The ability of BL2.2 to bind the same epitope on CTXB as the previously reported $\text{V}_\text{H}\text{H}$ construct BL3.2 was assessed using a competitive binding DELFIA similar to the methods described above.21 In short, 60 µl of BL3.2 (3.2 µM) was directly immobilized onto 96-well Immuno Plates (Thermo Scientific, 437111). BL2.2 (360 nM) was mixed with biotinylated CTXB (36 nM) and added to the plate. A biotinylated toxin-only control (36 nM) was included as well. Fluorescence was detected by adding 100 ng ml−1 of streptavidin-conjugated europium (PerkinElmer, 1244-360) diluted in DELFIA assay buffer (PerkinElmer, 4002-0010) and DELFIA enhancement solution 20 (PerkinElmer, 4001-0010), followed by intensity measurements using a Victor Nivo microplate reader. The blocking capacity was determined by relative comparison to the negative toxin-only control.
Detection of intracellular cAMP in cell-based assay
The ability of BL2.2 to neutralize the cytotoxic activity of CTX and LT was determined using a human HCA-7 colon cancer cell line (AddexBio, C0009003) as previously described.21 CTX (0.23 nM) or LT (4.60 nM) was incubated with different concentrations of either BL2.2 (0–62.5 nM and 0–78 nM, respectively) or a bivalent $\text{V}_\text{H}\text{H}$ construct without specificity for CTX or LT (57.5–230 nM and 1.15–4.60 µM, respectively) before being added to $1 \times 10^4$ cells per well. BL2.2 was analyzed in duplicates of technical triplicates, the bivalent $\text{V}_\text{H}\text{H}$ control was analyzed once in replicates of six. The amount of intracellular cAMP was detected after 2 hours according to the manufacturer’s protocol (cAMP-Glo™ Max Assay, Promega, Madison, WI). Luminescence was measured using a Victor Nivo Multimode plate reader.
The cAMP standard curve was prepared in triplicates according to the manufacturer’s protocol (cAMP-Glo™ Max Assay, Promega, Madison, WI) and the effect of BL2.2 on the concentration of intracellular cAMP determined using a sigmoidal four parameter logistic regression model ($R^2 = 0.9745$). The relative $\text{IC}_{50}$ for BL2.2 was calculated based on a variable slope model. Neutralization capacity of BL2.2 against CTX and LT in HCA-7 cells was calculated relative to toxin-only and untreated (HCA-7) controls using the following equation:
$$\text{Neutralization capacity} (\%) = 100 \times \frac{(\text{BL2.2 + Toxin}) -\text{Toxin}}{\text{HCA7untreated - Toxin}}$$
Structure prediction and characterization of the BL2.2-toxin interface
Artificial intelligence-based tools have revolutionized the field of protein structure prediction.33-36 In this study, ColabFold in combination with classical molecular dynamics (MD) simulations was used to predict and characterize the protein-protein interface of BL2.2 in complex with CTXB and LTB pentamers, respectively. Starting from the predicted complex structures, three repetitions of 1000 ns of classical MD simulations were performed, using the AMBER 24 simulation software containing the pmemd.cuda module.37 The structure models were placed into cubic water boxes of TIP3P water molecules with a minimum wall distance to the protein of 12 Å.38-40 Parameters for all simulations were derived from the AMBER force field 14SB.41 To neutralize the charges, we used uniform background charges.42-44 Each system was carefully equilibrated using a multistep equilibration protocol.45 Bonds involving hydrogen atoms were restrained using the SHAKE algorithm, allowing a timestep of 2.0 femtoseconds.46 System pressure was maintained at 1 bar by applying weak coupling to an external bath using the Berendsen algorithm.47 The Langevin thermostat was utilized to keep the temperature at 300 K during the simulations.48
The obtained MD simulations for BL2.2 in complex with CTXB and LTB pentamers were used to calculate contacts using the GetContacts software.49 The analysis performed using GetContacts software enabled the computation of interactions within a single protein structure, as well as between different protein interfaces, while also monitoring the evolution of contacts throughout the simulation. In addition to quantifying the contacts of the different poses, CPPTRAJ was utilized for hierarchical agglomerative clustering analysis to identify the most representative binding poses.50
Complex formation studies
Recombinant LT (15 μM) or CTX (68 μM) were mixed with $\text{V}_\text{H}\text{H}$ constructs at a 2:1 (BL2.1:toxin) or 1:1 (BL2.2:toxin) molar ratio, to equalize the number of ($\text{V}_\text{H}\text{H}$ binding sites. Samples were analyzed by SEC on an equilibrated (PBS) Superdex 200 increase 10/300 GL column (Cytiva) using the ÄKTA Pure system (GE Healthcare). Chromatograms were normalized to the maximum peak absorbance and compared to the SEC profiles of LT, CTX, BL2.2, or BL2.1 alone.
Protein deglycosylation
BL2.2 and the previously characterized CTXB-specific bivalent $\text{V}_\text{H}\text{H}$ construct were diluted to $1\,\text{mg}\,\text{ml}^{-1}$ in $0.1\,\text{M}\,\text{KH}_2\text{PO}_4$ (pH 5.5) and mixed with endoglycosidase H (Sigma-Aldrich, 324717) at a 100:1 volume ratio. The mixtures were incubated at 25 °C for 2 hours and deglycosylation confirmed by SDS-PAGE (Fig. S1).
ETEC colonization in a Caco-2 intestinal flow chamber model
The intestinal epithelium colonization model was a modified version of a previously published model.51 Caco-2 cells (ATCC, HTB-37) were maintained in Dulbecco's Modified Eagle's Medium (Gibco, 31966-021) enriched with 20% heat-inactivated fetal bovine serum (HI-FBS) (Biowest, S181H) and 1% penicillin-streptomycin (Corning, 30-002-CI). Cells were passaged at 60-90% confluency and used for experiments between passages 25 and 50. All cultures were maintained at 5% $\text{CO}_2$ and 37 °C. For flow chamber culturing, cells were seeded in collagen-coated μ-slides (Ibidi, 80606) at a density of $1.2 \times 10^5$ cells cm-2 and flow chambers subsequently connected to silicon tubing and an IsmatecTM IPC High Precision Multichannel Pump. Cells were allowed to settle and adhere for 4 hours before initiating flow at a rate of $15\,\mu\text{l}\,\text{min}^{-1}$ (Shear-stress: $0.019\,\text{dyn}\,\text{cm}^{-2}$). One day post-seeding (DPS), the flow rate was increased to $60\,\mu\text{l}\,\text{min}^{-1}$, and, at 2 DPS, a pulsatile flow was applied ($140\,\mu\text{l}\,\text{min}^{-1}$, Shear-stress: $0.178\,\text{dyn}\,\text{cm}^{-2}$) for 1 minute ON and 9 minutes OFF to simulate the intermittent nature of peristaltic movement in the gut. At 6 DPS, the medium was replaced with serum-reduced medium (2% HI-FBS) without antibiotics. This was done with or without the addition of $2.2\,\mu\text{g}\,\text{ml}^{-1}$ of BL2.2, and the cells were incubated for 30 minutes at flow rate of $140\,\mu\text{l}\,\text{min}^{-1}$ prior to infection with ETEC H10407 or LT-deficient mutant H10407ΔeltA (jf570) for 24 hours.52-54
Inocula were prepared by suspending bacteria from agar plates in Hank's Balanced Salt Solution (HBSS) (Gibco, 14025092) to an OD of 0.2, corresponding to an inoculum of approximately $1.3 \times 10^8$ CFU ml-1. Inocula were introduced under a flow of $15\,\mu\text{l}\,\text{min}^{-1}$ for 10 min, before shifting to sterile media with a pulsatile flow (90 μl min-1, 1 minute ON, 9 minutes OFF) for the remaining infection period. ETEC-infected cell layers were harvested using 0.25% trypsin-EDTA (Biowest, L0932) with the addition of 0.1% Triton X-100 (Sigma-Aldrich, X100), and plated on Luria Bertani (LB) agar plates for estimation of colony-forming units (CFUs).
For intestinal cell viability analysis, Caco-2 cell layers within the μ-slides were stained using the LIVE/DEAD™ Viability/Cytotoxicity Kit (Thermo Fisher Scientific, L3224) with a modified protocol. Briefly, the cells were washed with HBSS, stained with 2 μM Calcein-AM for 30 minutes, rinsed again with HBSS, and then fixed with formalin for 15 minutes. To visualize tight junctions, cell layers were washed with HBSS, fixed with 10% formalin (Sigma-Aldrich, HT501128) for 15 minutes, and and rinsed with PBS. Permeabilization was performed using 0.1% Triton X-100 for 15 minutes, followed by additional PBS washes. Blocking was carried out for 60 minutes with 5% bovine serum albumin (BSA) (Sigma Aldrich, A9418). Cells were incubated for 3 hours at room temperature with $2\,\mu\text{g}\,\text{ml}^{-1}$ Occludin Monoclonal Antibody Alexa Fluor 488 (Invitrogen, OC-3F10) in 1% BSA followed by counterstaining with 500 nM 4′,6-diamidino-2-phenylindole (DAPI) for 10 minutes (Sigma-Aldrich, D9542). Fluorescence microscopy was carried out on an Olympus BX51 fluorescence microscope, equipped with a 10x objective lens and using Olympus cellSens Dimensions software (version 1.7).
Data processing and visualization
GraphPad Prism (version 10) and R (version 4.3.2) were used for figure generation and statistical analyzes. For CFU comparisons, one-way ANOVA with Tukey's HSD post-hoc test was performed using the aov() and TukeyHSD() function from the R stats package (version 4.3.2) (***: p$ < 0.001$, **: p$ < 0.01$, *: p$ < 0.05$). Average values and standard deviations were calculated after transforming the values to the figure scale illustrated. CLC Main Workbench version 23.0.2 was used for sequence analysis and alignment.
Results
The monovalent BL2.1 binds to LT and CTX with high affinity
When characterizing an antibody construct for its ability to displace the natural interaction between a toxin and its receptor, binding affinity is a critical parameter. LTB and CTXB pentamers have apparent affinities in the low nanomolar range (0.57 and 0.73 nM, respectively) for multivalent binding to the GM1 receptor, due to the avidity effect.13 BLI and SPR were used to determine the binding affinity of BL2.1, the monovalent counterpart of BL2.2, towards the LTB- and CTXB-pentamers. BLI measurements of immobilized monovalent $\text{V}_\text{H}\text{H}$ BL2.1 against LTB and CTXB pentamers demonstrated apparent affinities of < 0.001 nM and 0.33 nM, respectively (Table S1 and Fig. S2). SPR analysis of the same ligand–analyte interactions (BL2.1–LTB/CTXB) yielded affinities (avidities) of 0.003 nM for LTB and 0.37 nM for CTXB pentamers, respectively (Table S1 and Fig. S3). SPR analysis of the opposite biomolecular interaction (i.e., LTB and CTXB pentamers as ligands), revealed a BL2.1 binding affinity of 0.26 nM towards LTB and 8.23 nM towards CTXB (Table S1 and Fig. S4).
BL2.2 inhibits CTX functionality
Previous studies have shown that BL2.1 (i.e., LT109) can bind both LT and CTX, and hinder the LT-GM1 interaction.30 Additionally, its bivalent counterpart, BL2.2 ($\text{V}_\text{H}\text{H}$–$\text{(GGGGS)}_3$–$\text{V}_\text{H}\text{H}$), has been shown to bind and neutralize LT from porcine ETEC strains in vivo.26,55 In a DELFIA,we specifically tested the in vitro CTXB-GM1 blocking capacity of BL2.1 and BL2.2. Both the monovalent BL2.1 and the bivalent BL2.2 blocked 100% of the interaction between CTXB and GM1 at a 1:1 CTXB:$\text{V}_\text{H}\text{H}$/$\text{V}_\text{H}\text{H}$-$\text{V}_\text{H}\text{H}$ molar ratio of binding sites, comparable to their blocking capacity (100%) of LTB–GM1 interaction at identical ratios (Figure 1). In contrast, a previously reported anti-CTX $\text{V}_\text{H}\text{H}$ selected based on target binding ability, demonstrated a CTXB-GM1 blocking capacity of 19% at a 1:100 toxin:$\text{V}_\text{H}\text{H}$ ratio without the ability to hinder LTB–GM1 interaction at the same concentration (Fig. S5).56,57 In the remainder of the study, the bivalent format of BL2.2 was employed to exploit avidity, given the multivalent nature of the LTB/CTXB pentamer.21,58,59
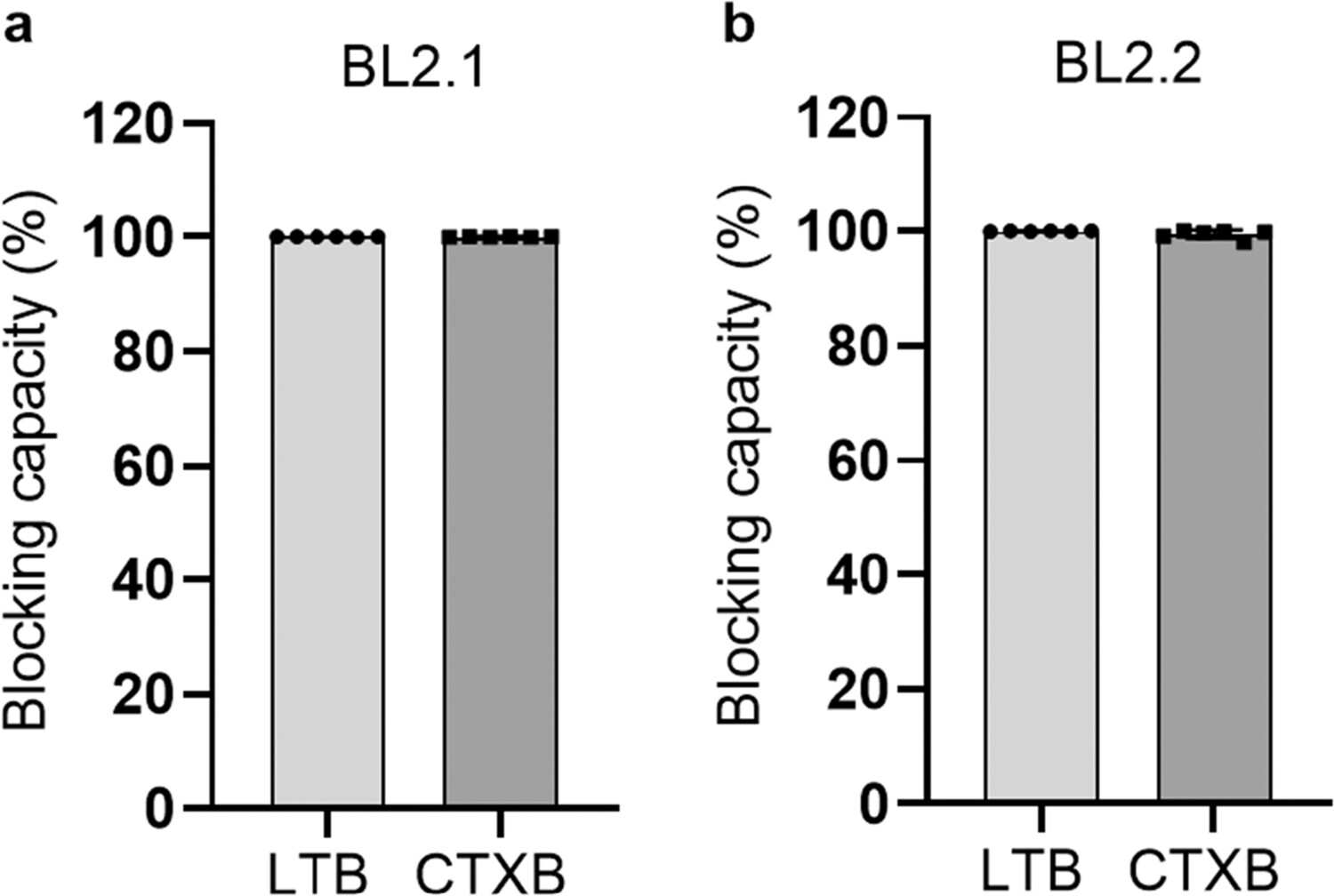
Figure 1. BL2.2 blocking capacity of LTB– and CTXB–GM1 interaction. (a) The ability of the monovalent $\text{V}_\text{H}\text{H}$ BL2.1 to abrogate the interaction between LTB and CTXB pentamers with the intestinal ganglioside receptor GM1 at a molar ratio of 1:1 (binding sites of LTB/CTXB/$\text{V}_\text{H}\text{H}$). The average blocking capacity was calculated by normalization against a toxin-only control, after duplicate measurements of technical triplicates. Error bars indicate standard deviation. (b) The ability of $\text{V}_\text{H}\text{H}$-$\text{V}_\text{H}\text{H}$ BL2.2, the bivalent counterpart of BL2.1, to block toxin (LTB or CTXB) interaction with GM1 at a molar ratio of 1:1 (LTB/CTXB/$\text{V}_\text{H}\text{H}$ binding sites). The average blocking capacity was calculated by normalization against a toxin-only control, after duplicate measurements of technical triplicates. Error bars indicate standard deviation.
BL2.2 neutralizes LT- and CTX-induced cytotoxicity
Uptake of LT and CTX by endothelial cells of the small intestine induces increased levels of cellular cAMP.18,60 The toxin-neutralizing capacity of BL2.2 was assessed by adding increasing concentrations of BL2.2 (0–78 nM) to LT (4.60 nM) or 0–62.5 nM BL2.2 to CTX (0.23 nM) and measuring the intracellular (HCA-7) cAMP concentration. BL2.2 reduced intracellular cAMP levels in HCA-7 intestinal epithelial cells seven-fold for LT and three-fold for CTX, relative to toxin alone (Figure 2). Normalized to this baseline, BL2.2 fully blocked cellular uptake of both LT and CTX at molar ratios of 1:2 and 1:135 (toxin:BL2.2), respectively (Fig. S6). This corresponds to a 5:4 (LT:BL2.2) and 5:270 (CTX:BL2.2) binding site ratio. By comparison, GM1-coated nanoparticles—used as receptor decoys to inhibit the CTX–GM1 interaction—achieved only a three-fold cAMP reduction in HCA-7.60 Notably, a bivalent $\text{V}_\text{H}\text{H}$ lacking toxin specificity showed no measurable impact on LT or CTX uptake (Fig. S6).
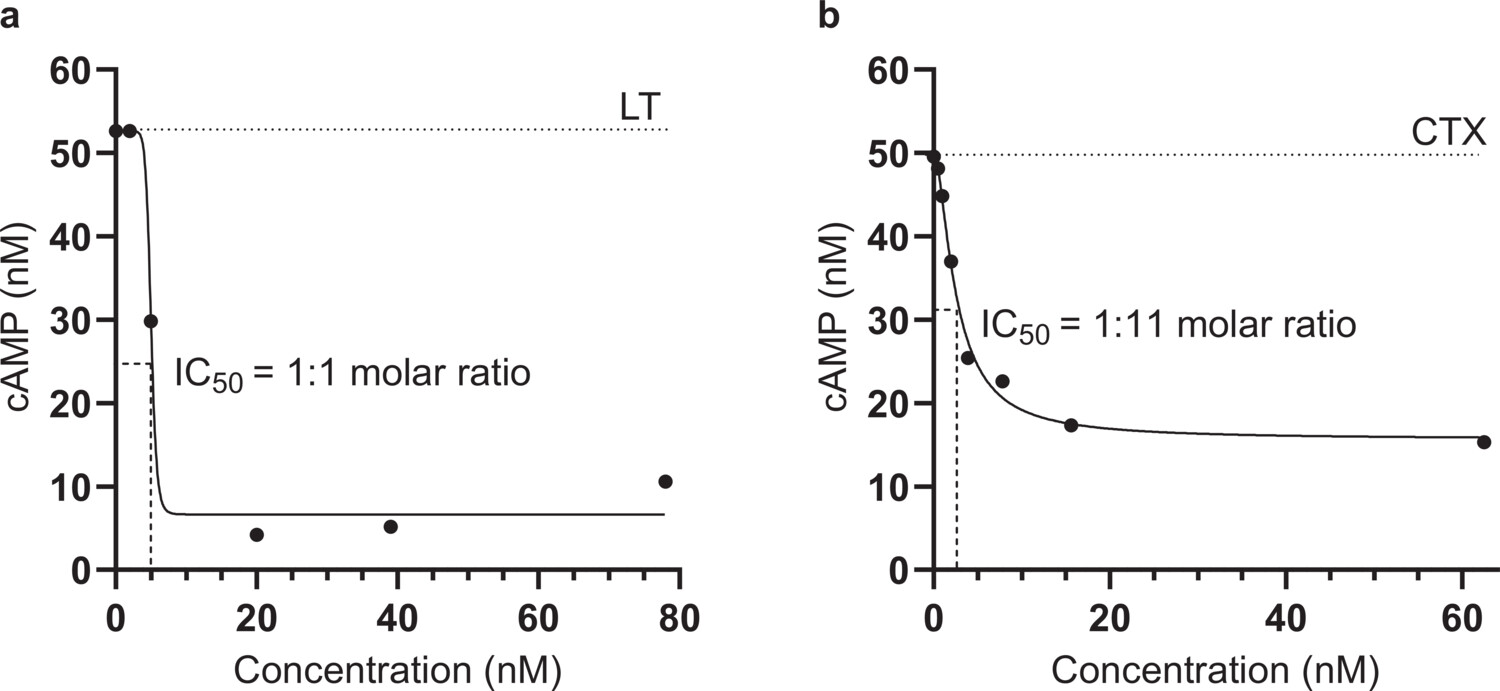
Figure 2. BL2.2 neutralization of LT and CTX toxicity in a human cell-based assay. (a) Neutralization of LT (4.60 nM) cytotoxicity in the presence of increasing concentrations (0–78 nM) of BL2.2. Levels of intracellular cAMP were interpolated from a sigmoidal four parameter logistic cAMP standard curve ($R^2 = 0.9745$) based on triplicate measurements. Each data point represents an interpolated mean value from biological duplicates comprised of three technical replicates. (b) Neutralization of CTX (0.23 nM) cytotoxicity in the presence of increasing concentrations (0–62.5 nM) of BL2.2. Levels of intracellular cAMP were interpolated from a sigmoidal four parameter logistic cAMP standard curve ($R^2 = 0.9745$) based on triplicate measurements. Each data point represents an interpolated mean value from biological duplicates comprised of three technical replicates.
BL2.2 is predicted to bind to the conserved GM1 binding site
MD simulations were used to predict the most probable and stable complex formation between BL2.2 and both LT and CTX, respectively.21 Analysis of the protein–protein interface between BL2.2 and LT (Figure 3a) identified 11 distinct amino acids (E11, H13, N14, K34, R35, S55, Q56, H57, N90, W88, and K91) in the LTB epitope (Figure 3b). Among these residues, residue 13 is the only one that differs between LT and CTX in the GM1 binding site, with the exception of certain human LT (hLT) strains that carry an Arg at this position.15 Notably, S55 and K91 from two adjacent LTB subunits in the pentamer were predicted to contribute to the interaction. Ten amino acid residues (D31, D55, D99, D103, Y104, V105, S106, N108, E110, and T111) in the BL2.2 paratope are likely essential for LTB binding (Figure 3c). Analysis of the predicted BL2.2–CTX interaction (Figure 3d) revealed that out of the 11 amino acid residues (E11, R13, K34, R35, E51, S55, Q56, H57, Q61, W88, and K91) in the CTXB epitope, eight are shared by LTB (nine, if residue 13 is also considered) (Figure 3e). In this case, three residues (S55, Q56 and K91) contributed twice, from adjacent CTXB subunits. Similarly, eight out of the ten amino acid residues (D55, D99, S100, Y101, D103, Y104, V105, S106, E110, and T111) in the BL2.2 paratope important for CTXB binding, are shared between the two interactions (Figure 3f). Notably, the shared LTB/CTXB epitope of BL2.2 contains 9 of the 12 amino acids found in the CTXB epitope of BL3.2, a previously characterized $\text{V}_\text{H}\text{H}$ construct against CTX.21 Both BL2.2 and BL3.2 epitopes were predicted using equivalent MD simulations, with the BL3.2 epitope partially validated through hydrogen-deuterium exchange mass spectrometry (HDX-MS).21 In a competitive binding assay, BL2.2 prevented BL3.2 from binding CTXB at equimolar ratios ($\text{V}_\text{H}\text{H}$ construct:CTXB) (Fig. S7). The predicted BL2.2 epitope is in strong agreement with the established GM1 binding site found for both LT and CTX, greatly conserved among hLTB and porcine LTB (pLTB), as well as the ctxB genotypes (ctxB1, ctxB3, and ctxB7) associated with all cholera pandemics to date (Fig. S8).14,61
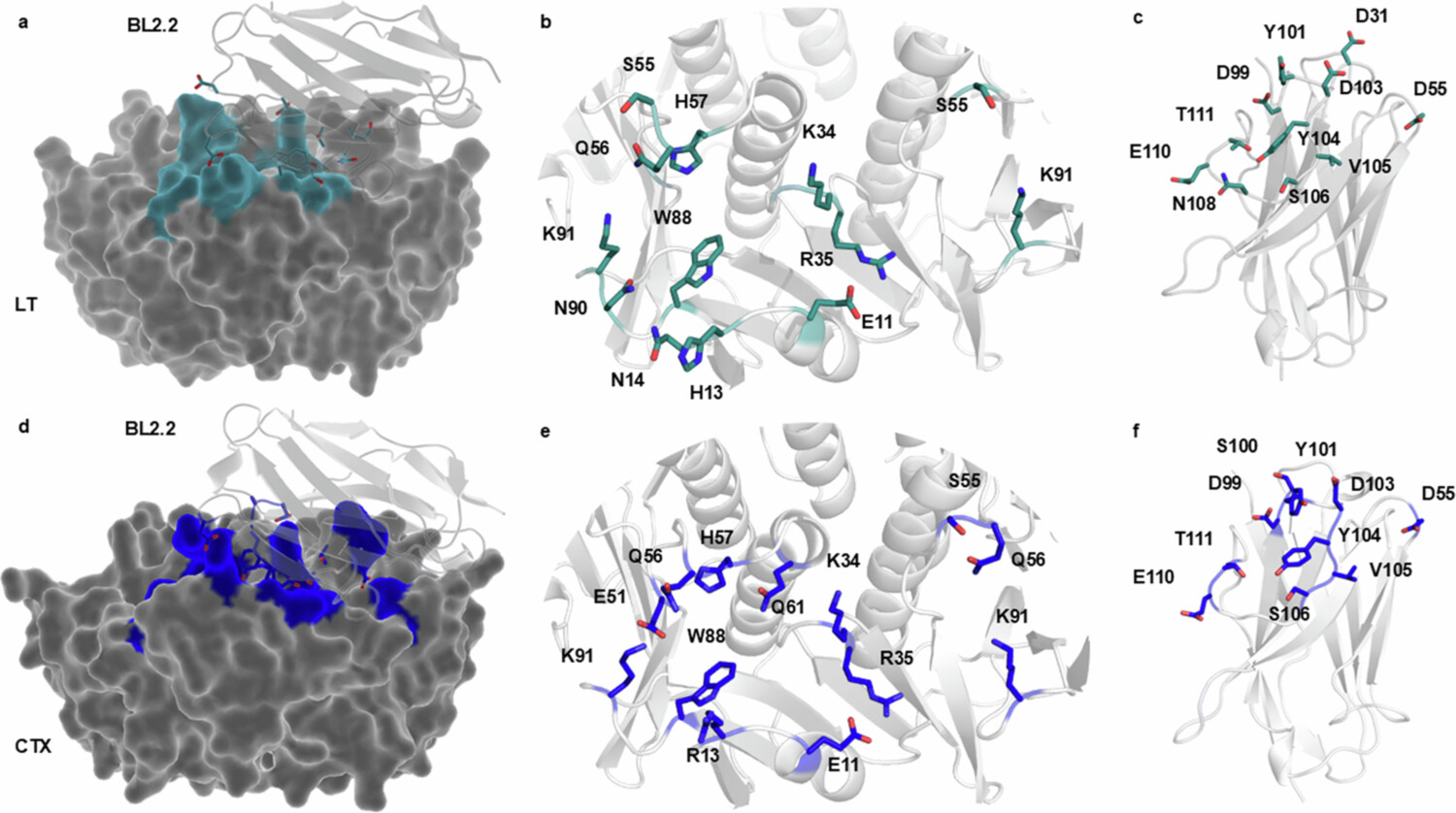
Figure 3. Prediction of the BL2.2 binding interface with LTB and CTXB pentamers. (a) The predicted protein-protein interface (blue) between BL2.2 (light gray) and LT (dark gray) based on ColabFold and molecular dynamics simulations. For visualization purposes, only one $\text{V}_\text{H}\text{H}$ of the bivalent BL2.2 molecule is shown. (b) The 11 amino acid residues (E11, H13, N14, K34, R35, S55, Q56, H57, W88, N90, and K91) in the LTB epitope predicted to be essential for BL2.2 binding, including three residues (S55, Q56, and K91) from the adjacent subunit in the pentamer. (c) The 11 amino acid residues (D31, D55, D99, Y101, D103, Y104, V105, S106, N108, E110, and T111) in the BL2.2 paratope predicted to be crucial for LT interaction. (d) The predicted protein-protein interface (cyan) between BL2.2 (light gray) and CTX (dark gray) based on ColabFold and molecular dynamics simulations. For visualization purposes, only one $\text{V}_\text{H}\text{H}$ of the bivalent BL2.2 molecule is shown. (e) The 11 amino acid residues (E11, R13, K34, R35, E51, S55, Q56, H57, Q61, W88, and K91) in the CTXB epitope predicted to be essential for BL2.2 binding, including three residues (S55, Q56, and K91) from the adjacent subunit in the pentamer. (f) The 10 amino acid residues (D55, D99, S100, Y101, D103, Y104, V105, S106, E110, and T111) in the BL2.2 paratope predicted to be important for CTXB pentamer interaction.
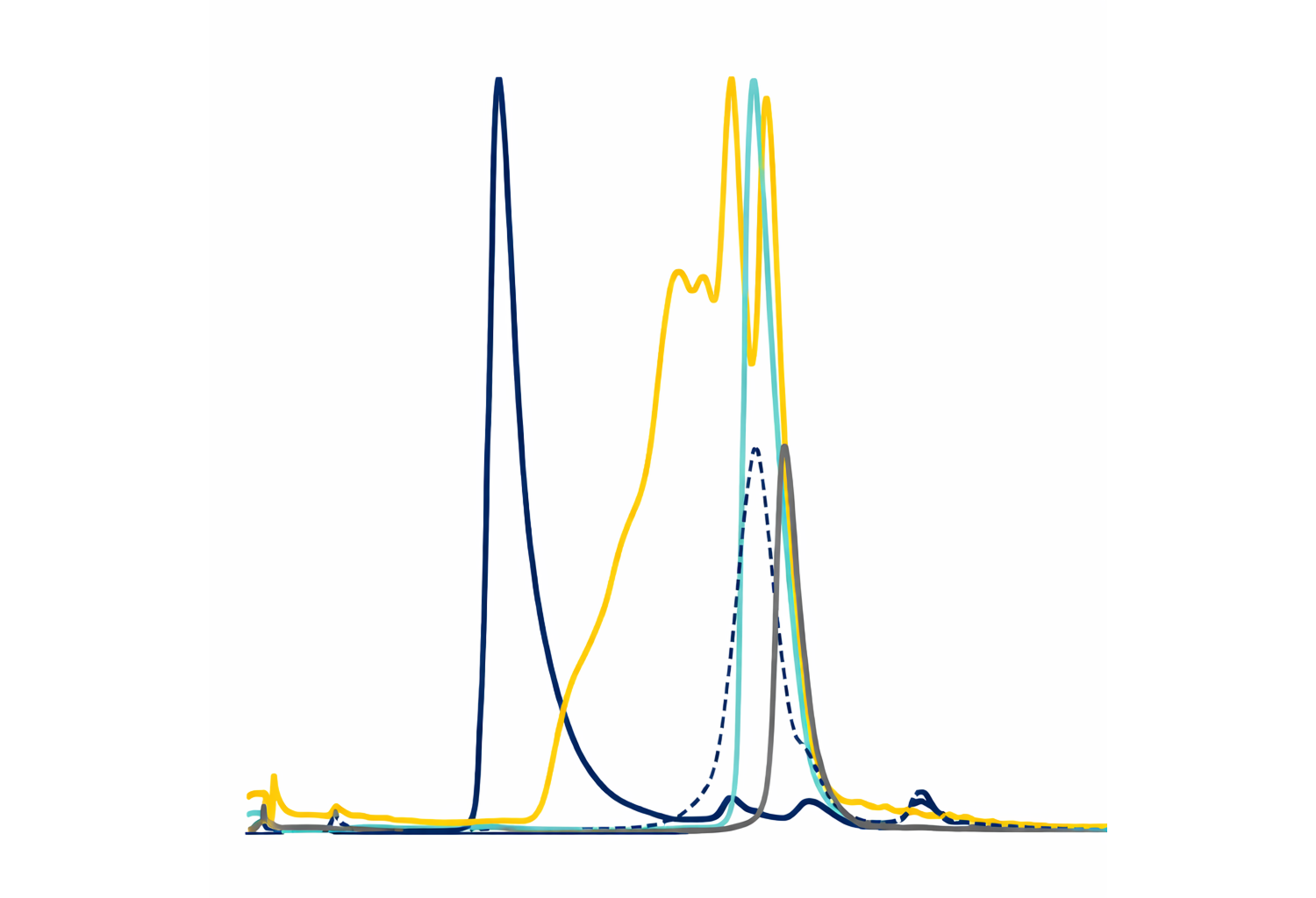
BL2.2 promotes toxin aggregation for enhanced neutralization
The SEC profile of BL2.2 incubated with LT or CTX showed a peak eluting close to the void volume of the column, indicating that the bivalent $\text{V}_\text{H}\text{H}$ construct induced toxin aggregation (Figure 4a, b). In contrast, incubating the toxins with the monovalent BL2.1 at equivalent binding site ratios resulted in smaller protein complexes.
The original study of BL2.1 (LT109) reported that N-glycosylation enhances LT neutralization, potentially by increasing steric hindrance.30 Here, SPR analysis of LTB and CTXB pentamer interaction with glycosylated and non-glycosylated BL2.2 demonstrated that the N-glycans increased antigen avidity (Table 1, Fig. S9, and Fig. S10). N-glycosylated BL2.2 exhibited higher avidity towards LTB (0.27 nM) and CTXB (5.15 nM) pentamers compared to the non-glycosylated counterpart (1.25 nM and 8.55 nM, respectively).
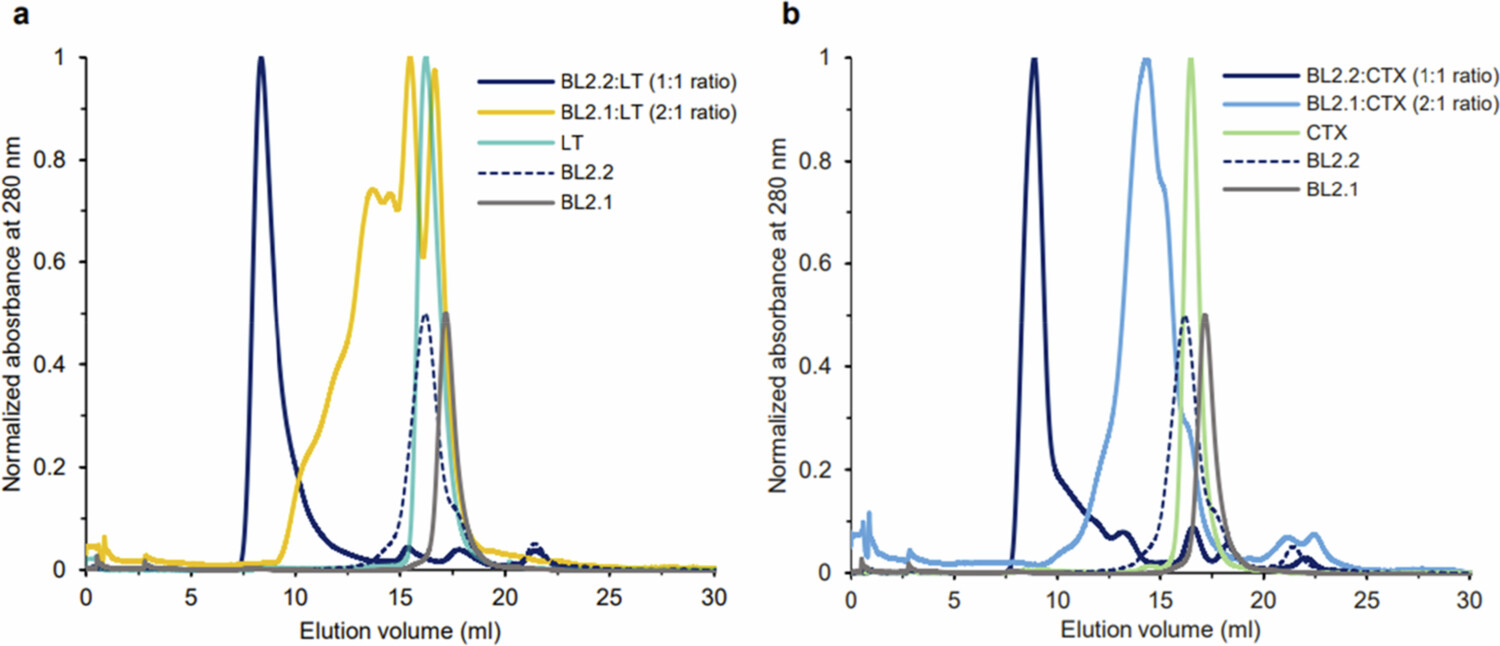
Figure 4. Size-exclusion chromatography (SEC) analysis of BL2.2 and BL2.1 complex formation with LT or CTX holotoxins. (a) Superimposition of SEC profiles of LT in complex with the bivalent $\text{V}_\text{H}\text{H}$ BL2.2 (dark blue) or its monovalent counterpart BL2.1 (yellow) with molar ratios indicated. For comparison, chromatograms of LT alone (cyan), BL2.2 (dashed blue line), and BL2.1 (gray) are also included. (b) Superimposition of SEC profiles of CTX in complex with the bivalent $\text{V}_\text{H}\text{H}$ BL2.2 (dark blue) or its monovalent counterpart BL2.1 (light blue). Profiles of CTX alone (green), BL2.2 (dashed blue line), and BL2.1 (gray) are also displayed. To aid in visual presentation, the absorbance of the $\text{V}_\text{H}\text{H}$ constructs alone was reduced to half in both panels.
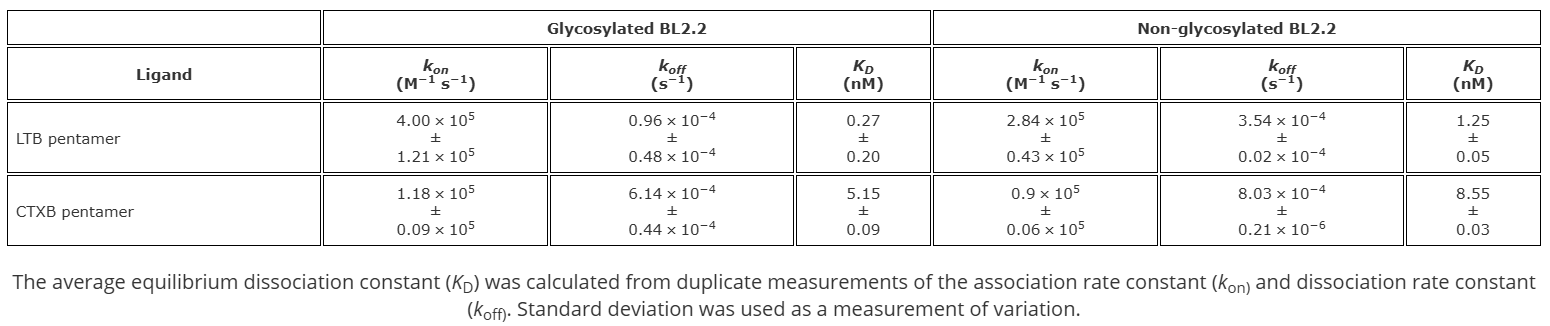
Table 1. Comparison of N-glycosylated and non-glycosylated BL2.2 binding avidity towards LTB and CTXB pentamers determined using surface plasmon resonance (SPR).
BL2.2 abrogates ETEC virulence in a Caco-2 flow chamber infection model
LT has been reported to promote adhesion of ETEC to intestinal epithelial cells.62,63 In this study, an intestinal flow chamber infection model was utilized to evaluate the effect of LT neutralization by BL2.2 on ETEC virulence.51 Caco-2 cell layers were infected with either ETEC H10407 or an LT-deficient isogenic mutant (H10407ΔeltA), which exhibits reduced adhesion and impaired intestinal colonization.62,64 The LT-deficient mutant (H10407ΔeltA) was used to evaluate whether neutralization of the LTB pentamer by BL2.2 causes ETEC H10407 to behave as an LT-deficient strain.
CFU counts from infected cell layers revealed a significant ($p = 4.2 \times 10^{-6}$) 10-fold reduction in H10407 colonization after 24 hours when BL2.2 ($2.21\,\mu\text{g}\,\text{ml}^{-1}$) was added to the medium (Figure 5a). The impact of BL2.2 on ETEC colonization was more pronounced than the effect observed for the LT-deficient mutant (H10407ΔeltA), but this difference was not statistically significant (Figure 5a,b). BL2.2 did not significantly affect colonization by the LT-deficient mutant (H10407ΔeltA), supporting that BL2.2 neutralizes LT specifically. Tight junction integrity was compromised in H10407-infected cell layers, as evidenced by a more diffuse distribution of occludin staining, compared to layers co-incubated with BL2.2 or infected with the H10407ΔeltA mutant. Similarly, cell viability, assessed using Calcein-AM staining, was significantly reduced in H10407-infected layers (Fig. S11).
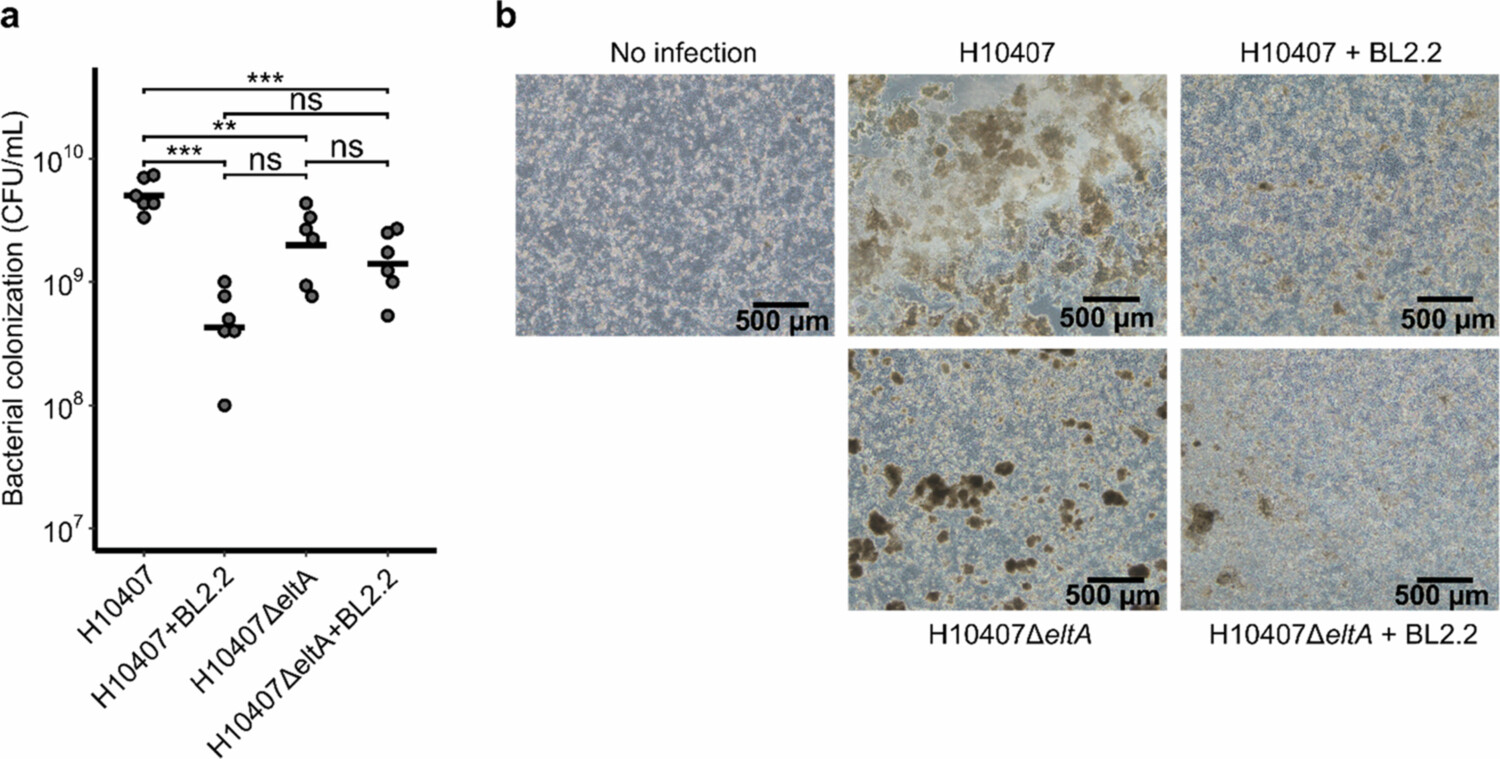
Figure 5. Suppression of ETEC colonization and virulence by BL2.2 in an intestinal flow chamber infection model. (a) CFU counts (CFU ml-1) after 24 hours infection in Caco-2 flow chambers: ETEC H10407 with BL2.2 ($p = 4.2 \times 10^{-6}$), the LT-deficient mutant H10407ΔeltA with BL2.2 ($p = 1.4 \times 10^{-6}$), and the LT-deficient mutant H10407ΔeltA without BL2.2 ($p = 1.9 \times 10^{-3}$), compared to ETEC H10407 alone. Statistical analyzes were performed using a one-way ANOVA with Tukey's HSD post-hoc test (***: $p < 0.001$, **: $p < 0.01$, *: $p < 0.05$). (b) Phase contrast microscope images of infected Caco-2 cell layers, the same used for CFU counts in panel (a). Bacterial aggregates are visible on the surface of the Caco-2 cell layers.
Discussion
ETEC and V. cholerae are endemic to resource-limited settings, and their co-infection prevalence has been on the rise during the last decades.65 Oral cholera vaccines (OCVs) have been instrumental in combating cholera-induced diarrhea and Dukoral, which combines inactivated V. cholerae with 1 mg of recombinant CTXB, is the only licensed vaccine with a substantial effect (67% protection three months post-administration) also against ETEC diarrhea.66-70 Recent ETEC-vaccine development efforts have focused on combining CFA and recombinant toxoids, such as LTB, CTXB, or LTB–CTXB hybrids to raise robust mucosal antibody responses and achieve broad strain coverage.71,72 However, while a broadly protective ETEC vaccine offers the potential for extensive protection, the associated higher development and implementation costs present significant barriers to accessibility and affordability in LMICs.8 Ultimately, effective population protection using enteric vaccines in resource-limited settings is complicated by poor distribution infrastructure, their impaired protection (e.g., due to environmental enteropathy), and a lack of financial incentives.73-77
Here, we evaluate a previously reported bivalent $\text{V}_\text{H}\text{H}$ construct BL2.2 based on its ability to neutralize both LT and CTX functionality in vitro and protect Caco-2 cells from ETEC (H10407 $\text{LT}^+$ $\text{ST}^+$) infection.26 BL2.2 was able to effectively hinder cellular uptake of both LT and CTX similar to a previously characterized $\text{V}_\text{H}\text{H}$ construct (i.e., BL3.2) that significantly decrease intestinal fluid secretion and burden of V. cholerae.21 Moreover, BL2.2 limited ETEC pathogenesis by significantly reducing bacterial colonization and preserving host cell viability and barrier integrity in an intestinal flow chamber model. This effect appears to be associated with the ability of BL2.2 to efficiently aggregate LT, preventing its interaction with host cells and thereby inhibiting the pathogenic effects of this toxin. Aggregation is likely induced by BL2.2 simultaneously engaging two B-pentamers, thereby cross-linking the toxins. This aligns with previous findings from other groups, which demonstrated that mismatched valency between toxins and inhibitors promotes aggregation and enhances potency.78-80 Moreover, the $\text{V}_\text{H}\text{H}$ construct bind to two adjacent subunits in LTB or CTXB pentamers, which could further enhance the interaction.
The reduced colonization observed upon LT neutralization corroborates previous findings that LT promotes adherence of ETEC.62,63 In ETEC strains expressing F4 fimbriae, this mechanism is thought to involve the ADP-ribosylation activity of LTA, which elevates host intracellular cAMP levels, thereby stimulating the production of bacterial adhesins and promoting colonization.63 The extent to which this effect on adhesion involves modulation of the major intestinal adhesion factor CFA/I in H10407 remains to be determined. Yet, these findings suggest that the ability of BL2.2 to neutralize LT might be sufficient to limit the multifaceted intestinal pathogenicity of the prototypical LT+ ST+ ETEC strain responsible for human infection. However, while the intestinal flow chamber model effectively simulates physiologically relevant shear stress, which influences bacterial pathogenesis and stimulates epithelial development, it does not replicate key human host factors, such as immune cell interactions, host microbiota, and complex biochemical gradients..81,82 The absence of these factors should also be considered when evaluating the ability of BL2.2 to mitigate ETEC virulence.
Similar to previous studies on V. cholerae pathogenicity, our findings indicate that a $\text{V}_\text{H}\text{H}$ against LT or CTX can eliminate the role of these toxins in pathogenesis.21,83 The use of cross-protective antibodies against LT and CTX at the mucosal surface of the GI tract has been explored by several novel vaccines.62,84-87 Vaccines targeting conserved antigens (e.g., LT or highly conserved CFs) aim to stimulate heterotypic immunity, providing broad protection across diverse strains. However, mucosal antibody immunity in the gut tends to be relatively short-lived without frequent re-exposure.85,88 In our earlier study of piglets subjected to ETEC challenge, two oral administrations of 12.1 mg of BL2.2 were given per day and supported a diverse gut microbiota.26 Similarly, daily administrations of an anti-rotavirus $\text{V}_\text{H}\text{H}$ ($35\,\text{mg}\,\text{kg}^{-1}$) have been shown to reduce diarrheal onset in small children.89 $\text{V}_\text{H}\text{H}$ constructs possess several biophysical properties (e.g., high thermal stability and long-term storage stability) that could make them well-suited for diarrheal disease management in LMIC settings.59,90 Recently, the BL2.2 construct demonstrated robust thermal and processing stability, maintaining structural integrity and LT-binding activity after lyophilization and exposure to elevated temperatures (up to 95 °C) and high humidity.55 Such resilience supports its potential suitability for storage, transport, and deployment in LMIC settings, where exposure to elevated temperatures and lack of reliable cold chain infrastructure are common challenges.91 Furthermore, reported yields of $\text{V}_\text{H}\text{Hs}$ reaching $31\,\text{g}\,\text{l}^{-1}$ in Komagataella phaffii or Aspergillus niger along with titers approaching $100\,\text{g}\,\text{l}^{-1}$ for similarly sized proteins (cellulases) in Trichoderma, suggest that production costs can be driven well below $\text{\$}$1 per day, assuming daily intake in the milligram range.92-94 This cost profile is highly favorable compared to conventional monoclonal antibodies ($ \text{\$} $95-200 per gram) and indicates that $\text{V}_\text{H}\text{H}$ constructs such as BL2.2 could provide affordable and continuous protection against enteric pathogens if consumed on a daily basis when at risk of infection.23,26,95
Half of all ETEC strains circulating in the human population produce LT, either alone or in combination with ST.96,97 The distribution of ETEC toxin profiles varies depending on geographical (e.g., seasonality and location) and host factors (e.g., age).98 Despite of the high diversity of ETEC strains found in patients, genomic analysis has inferred that ETEC-mediated diarrheal disease is most likely a set of overlapping global epidemics of individual ETEC lineages, which have been stable over considerable periods of time.99 This is deduced, as LT or ST gene alleles are linked to the chromosomal background. Toxin gene acquisition appears to provide a fitness advantage and remains key to virulence, as other E. coli strains can also acquire these genes to cause disease.100-102 In a comparable manner to LT, ST disrupt ion transport across intestinal epithelia, can cause watery diarrhea, and be pivotal to ETEC pathogenesis.103 Importantly, ST-only ETEC lineages are capable of causing severe diarrhea and the presence of both toxins can synergistically exacerbate diarrheal symptoms.104-106 The prevalence of ST-only strains and the phenotypic variation in toxin production emphasizes the need for evaluating BL2.2 against several clinically relevant strains to indicate its broadness of protection.71,107,108
LT and CTX both bind additional glycosylated structures, such as blood group antigens, beyond the GM1 ganglioside.15,16,109 In particular, fucosylation creates functional CTX receptors, providing evidence that these toxins can intoxicate intestinal cells through mechanisms that are not exclusively dependent on the GM1 ganglioside.15,97,110-113 Similarly, toxin binding to diverse glycoconjugates may explain why N-glycosylated BL2.2 binds more strongly to LTB and CTXB pentamers compared to deglycosylated BL2.2. In addition, glycosylation can enhance stability (e.g., protease resistance) with minimal safety risk for VHHs confined to the GI tract, as demonstrated in long-term oral exposure studies.114,115
In addition to its role in toxin-mediated pathogenesis, prior studies have identified LT as a key factor in inducing enteropathic alterations on enterocyte surfaces and enhancing ETEC colonization.18,116 However, the combined downstream effects of these enterotoxins on a population level, such as their contribution to disease symptoms via secondary intestinal bindings sites or to childhood malnutrition, remain largely unexplored.104,110,112,117,118 Smaller recombinant antibody formats, such as $\text{V}_\text{H}\text{Hs}$, can be selected using display technologies (e.g., phage display) to achieve high specificity and affinity for distinct epitopes, including toxins and cryptic or less accessible sites.119 We speculate that these properties could make $\text{V}_\text{H}\text{Hs}$ valuable tools for studying target-specific disease mechanisms, particularly those involving highly conserved virulence factors. Such investigations can provide insights into their roles in severe illness and their association with both short-term effects (e.g., diarrhea) and long-term outcomes (e.g., malnutrition and growth stunting).
While mortality from ETEC-associated diarrhea has decreased, incidence rate has not decreased comparably, and better interventions for preventing disease are needed.120 We suggest that targeted anti-virulence approaches might hold promise as new tools for managing bacterial enteric pathogens in the future, either as complementary strategies to vaccination programs or as standalone interventions for communities vulnerable to infection.
References
- Diarrheal disease deaths, by age. Our World in Data https://ourworldindata.org/grapher/deaths-from-diarrheal-diseases-by-age
- Causes of death in children under five. Our World in Data https://ourworldindata.org/grapher/causes-of-death-in-children-under-5
- 2018. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect Dis; 18:1211–1228.
- Nasrin S, Garbern SC, Gainey M, Kanekar S, Monjory M, Ahmed D, Qu K, Chu T, Schmid CH, Nelson EJ, et al. Clinical, sociodemographic and environmental risk factors for acute bacterial diarrhea among adults and children over five years in Bangladesh. Am J Trop Med Hyg. 2022;106:457–463. doi: 10.4269/ajtmh.21-0580.
- Mentzer A. von, Svennerholm A-M. Colonization factors of human and animal-specific enterotoxigenic Escherichia coli (ETEC). Trends Microbiol. 2024;32:448–464.
- Geurtsen J, de Been M, Weerdenburg E, Zomer A, McNally A, Poolman J. Genomics and pathotypes of the many faces of Escherichia coli. FEMS Microbiol Rev. 2022;46:fuac031. doi: 10.1093/femsre/fuac031.
- Frazão N, Sousa A, Lässig M, Gordo I. Horizontal gene transfer overrides mutation in Escherichia coli colonizing the mammalian gut. Proc Natl Acad Sci. 2019;116:17906–17915. doi: 10.1073/pnas.1906958116.
- Fleckenstein JM. Confronting challenges to enterotoxigenic escherichia coli vaccine development. Front Trop Dis. 2021;2. doi: 10.3389/fitd.2021.709907.
- Kansal R, Rasko DA, Sahl JW, Munson GP, Roy K, Luo Q, Sheikh A, Kuhne KJ, Fleckenstein JM, Payne SM. Transcriptional modulation of enterotoxigenic escherichia coli virulence genes in response to epithelial cell interactions. Infect Immun. 2013;81:259–270. doi: 10.1128/IAI.00919-12.
- Fleckenstein JM, Hardwidge PR, Munson GP, Rasko DA, Sommerfelt H, Steinsland H. Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes Infect. 2010;12:89–98. doi: 10.1016/j.micinf.2009.10.002.
- Yamamoto T, Nakazawa T, Miyata T, Kaji A, Yokota T. Evolution and structure of two ADP-ribosylation enterotoxins, Escherichia coli heat-labile toxin and cholera toxin. FEBS Lett. 1984;169:241–246. doi: 10.1016/0014-5793(84)80326-9.
- Svennerholm A-M. From cholera to enterotoxigenic Escherichia coli (ETEC) vaccine development. Indian J Med Res. 2011;133:188–194.
- MacKenzie CR, Hirama T, Lee KK, Altman E, Young NM. Quantitative analysis of bacterial toxin affinity and specificity for glycolipid receptors by surface plasmon resonance. J Biol Chem. 1997;272:5533–5538. doi: 10.1074/jbc.272.9.5533.
- Merritt EA, Sixma TK, Kalk KH, van Zanten BAM, Hol WGJ. Galactose-binding site in Escherichia coli heat-labile enterotoxin (LT) and cholera toxin (CT). Mol Microbiol. 1994;13:745–753. doi: 10.1111/j.1365-2958.1994.tb00467.x.
- Holmner Å, Mackenzie A, Ökvist M, Jansson L, Lebens M, Teneberg S, Krengel U. Crystal structures exploring the origins of the broader specificity of escherichia coli heat-labile enterotoxin compared to cholera toxin. J Mol Biol. 2011;406:387–402. doi: 10.1016/j.jmb.2010.11.060.
- Heggelund JE, Bjørnestad VA, Krengel U. Vibrio cholerae and Escherichia coli heat-labile enterotoxins and beyond. In J Alouf, D Ladant, M.R Popoff (Eds.), The Comprehensive Sourcebook of Bacterial Protein Toxins (Fourth Edition). 2015. pp. 195–229 Boston: Academic Press.
- Guichard A, Cruz-Moreno B, Aguilar B, van Sorge NM, Kuang J, Kurkciyan AA, Wang Z, Hang S, Pineton de Chambrun GP, McCole DF, et al. Cholera toxin disrupts barrier function by inhibiting exocyst-mediated trafficking of host proteins to intestinal cell junctions. Cell Host Microbe. 2013;14:294–305. doi: 10.1016/j.chom.2013.08.001.
- Sheikh A, Tumala B, Vickers TJ, Martin JC, Rosa BA, Sabui S, Basu S, Simoes RD, Mitreva M, Storer C, et al. Enterotoxigenic Escherichia coli heat-labile toxin drives enteropathic changes in small intestinal epithelia. Nat Commun. 2022;13:6886. doi: 10.1038/s41467-022-34687-7.
- Spangler BD. Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiol Rev. 1992;56:622–647. doi: 10.1128/mr.56.4.622-647.1992.
- Glass RI, Svennerholm A, Stoll BJ, Khan MR, Hossain KMB, Hug MI, Holmgren J. Protection against cholera in breast-fed children by antibodies in breast milk. N Engl J Med. 1983;308:1389–1392. doi: 10.1056/NEJM198306093082304.
- Petersson M, Zingl FG, Rodriguez-Rodriguez E, Rendsvig JKH, Heinsøe H, Wenzel Arendrup E, Mojica N, Segura Peña D, Sekulić N, Krengel U, et al. Orally delivered toxin–binding protein protects against diarrhoea in a murine cholera model. Nat Commun. 2025;16:2722. doi: 10.1038/s41467-025-57945-w.
- Petersson M, Thrane SW, Gram L, Muyldermans S, Laustsen AH. Orally delivered single-domain antibodies against gastrointestinal pathogens. Trends Biotechnol. 2023;41(7):875–886.
- Fiil BK, Thrane SW, Pichler M, Kittilä T, Ledsgaard L, Ahmadi S, Hermansen GMM, Jelsbak L, Lauridsen C, Brix S, et al. Orally active bivalent VHH construct prevents proliferation of F4+ enterotoxigenic Escherichia coli in weaned piglets. iSci. 2022;25 104003. doi: 10.1016/j.isci.2022.104003.
- Rodriguez Rodriguez ER, Nordvang RT, Petersson M, Rendsvig JKH, Arendrup EW, Fernández Quintero ML, Jenkins TP, Laustsen AH, Thrane SW. Fit-for-purpose heterodivalent single-domain antibody for gastrointestinal targeting of toxin B from Clostridium difficile. Prot Sci. 2024;33:e5035. doi: 10.1002/pro.5035.
- Saelens X, Schepens B. Single-domain antibodies make a difference. Science. 2021;371:681–682. doi: 10.1126/science.abg2294.
- Jenkins TP, Ács N, Arendrup EW, Swift A, Duzs Á, Chatzigiannidou I, Pichler M, Kittilä T, Peachey L, Gram L, et al. Protecting the piglet gut microbiota against ETEC-mediated post-weaning diarrhoea using specific binding proteins. NPJ Biofilms Microbiomes. 2024;10:1–15. doi: 10.1038/s41522-024-00514-8.
- Lasaro MAS, Rodrigues JF, Mathias-Santos C, Guth BEC, Régua-Mangia AH, Ferreira AJP, Takagi M, Cabrera-Crespo J, Sbrogio-Almeida ME. Production and release of heat-labile toxin by wild-type human-derived enterotoxigenic Escherichia coli. FEMS Immunol Med Microbiol. 2006;48:123–131. doi: 10.1111/j.1574-695X.2006.00134.x.
- Uesaka Y, Otsuka Y, Lin Z, Yamasaki S, Yamaoka J, Kurazono H, Takeda Y. Simple method of purification of Escherichia coli heat-labile enterotoxin and cholera toxin using immobilized galactose. Microb Pathog. 1994;16:71–76. doi: 10.1006/mpat.1994.1007.
- Mojica N, Kersten F, Montserrat-Canals M, Huhn III GR, Tislevoll AM, Cordara G, Teter K, Krengel U. Using vibrio natriegens for high-yield production of challenging expression targets and for protein perdeuteration. Biochemistry. 2024;63:587–598. doi: 10.1021/acs.biochem.3c00612.
- Harmsen MM, van Solt CB, Fijten HPD. Enhancement of toxin- and virus-neutralizing capacity of single-domain antibody fragments by N-glycosylation. Appl Microbiol Biotechnol. 2009;84:1087–1094. doi: 10.1007/s00253-009-2029-1.
- Martin CD, Rojas G, Mitchell JN, Vincent KJ, Wu J, McCafferty J, Schofield DJ. A simple vector system to improve performance and utilisation of recombinant antibodies. BMC Biotechnol. 2006;6:46. doi: 10.1186/1472-6750-6-46.
- Dawson RM. Characterization of the binding of cholera toxin to ganglioside GM1 immobilized onto microtitre plates. J Appl Toxicol JAT. 2005;25:30–38. doi: 10.1002/jat.1015.
- Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373:871–876. doi: 10.1126/science.abj8754.
- Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Žídek A, Bridgland A, Cowie A, Meyer C, Laydon A, et al. Highly accurate protein structure prediction for the human proteome. Nature (London). 2021;596:590–596. doi: 10.1038/s41586-021-03828-1.
- Wohlwend J, Corso G, Passaro S, Reveiz M, Leidal K, Swiderski W, Portnoi T, Chinn I, Silterra J, Jaakkola T, et al. (2024) Boltz-1: Democratizing Biomolecular Interaction Modeling. 2024.11.19.624167 Preprint at. doi: 10.1101/2024.11.19.624167
- Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, Ronneberger O, Willmore L, Ballard AJ, Bambrick J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature (London). 2024;630:493–500. doi: 10.1038/s41586-024-07487-w.
- D.A. C, et al. Amber 2024. 2024. San Fransisco: University of California.
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. doi: 10.1063/1.445869.
- El Hage K, Hédin F, Gupta PK, Meuwly M, Karplus M. Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size. eLife. 2018;7:e35560. doi: 10.7554/eLife.35560.
- Gapsys V, de Groot BL. Comment on ‘valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size’. eLife. 2019;8:e44718. doi: 10.7554/eLife.44718.
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. doi: 10.1021/ja955032e.
- Darden T, York D, Pedersen L. Particle meshewald: an N⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. doi: 10.1063/1.464397.
- Salomon-Ferrer R, Case DA, Walker RC. An overview of the amber biomolecular simulation package. WIREs Comput Mol Sci. 2013;3:198–210. doi: 10.1002/wcms.1121.
- Hub JS, de Groot BL, Grubmüller H, Groenhof G. Quantifying artifacts in Ewald simulations of inhomogeneous systems with a net charge. J Chem Theory Comput. 2014;10:381–390. doi: 10.1021/ct400626b.
- Wallnoefer HG, Handschuh S, Liedl KR, Fox T. Stabilizing of a Globular protein by a highly complex water network: a molecular dynamics simulation study on factor Xa. JPCB. 2010;114:7405–7412. doi: 10.1021/jp101654g.
- Andersen HC. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. JCoPh. 1983;52:24–34. doi: 10.1016/0021-9991(83)90014-1.
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. doi: 10.1063/1.448118.
- Adelman SA, Doll JD. Generalized Langevin equation approach for atom/solid-surface scattering: General formulation for classical scattering off harmonic solids. J Chem Phys. 1976;64:2375–2388. doi: 10.1063/1.432526.
- Stanford University. GetContacts. https://GetContacts.github.io/index.html
- Roe DR, Cheatham TEI. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p.
- Pedersen RM, Grønnemose RB, Stærk K, Asferg CA, Andersen TB, Kolmos HJ, Møller-Jensen J. A method for quantification of epithelium colonization capacity by pathogenic bacteria. Front Cell Infect Microbiol. 2018;8:16. doi: 10.3389/fcimb.2018.00016.
- Dorsey FC, Fischer JF, Fleckenstein JM. Directed delivery of heat-labile enterotoxin by enterotoxigenic Escherichia coli. Cell Microbiol. 2006;8:1516–1527. doi: 10.1111/j.1462-5822.2006.00736.x.
- Chutkan H, Kuehn MJ. Context-dependent activation kinetics elicited by soluble versus outer membrane vesicle-associated heat-labile enterotoxin. Infect Immun. 2011;79:3760–3769.
- Mudrak B, Rodriguez DL, Kuehn MJ. Residues of heat-labile enterotoxin involved in bacterial cell surface binding. J Bacteriol. 2009;191:2917–2925. doi: 10.1128/JB.01622-08.
- Xu J, Andrani M, Kjærup RB, Dalgaard TS, Eriksen C, Laustsen AH, Brix S, Thrane SW, Canibe N. In-feed provision of binding proteins sustains piglet gut health and mitigates ETEC-induced post-weaning diarrhea. J Anim Sci Biotechnol. 2025;16:78. doi: 10.1186/s40104-025-01209-6.
- Goldman ER, Anderson GP, Liu JL, Delehanty JB, Sherwood LJ, Osborn LE, Cummins LB, Hayhurst A. Facile generation of heat stable antiviral and antitoxin single domain antibodies from a semi-synthetic llama library. AnaCh. 2006;78:8245–8255. doi: 10.1021/ac0610053.
- Legler PM, Zabetakis D, Anderson GP, Lam A, Hol WGJ, Goldman ER. Structure of a low-melting-temperature anti-cholera toxin: llama VHH domain. Acta Crystallograph Sect F Struct Biol Cryst Commun. 2013;69:90–93. doi: 10.1107/S1744309112050750.
- Hanke L, Das H, Sheward DJ, Perez Vidakovics L, Urgard E, Moliner-Morro A, Kim C, Karl V, Pankow A, Smith NL, et al. A bispecific monomeric nanobody induces spike trimer dimers and neutralizes SARS-CoV-2 in vivo. Nat Commun. 2022;13:155. doi: 10.1038/s41467-021-27610-z.
- Harmsen MM, De Haard HJ. Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol. 2007;77:13–22. doi: 10.1007/s00253-007-1142-2.
- Das S, Angsantikul P, Le C, Bao D, Miyamoto Y, Gao W, Zhang L, Eckmann L, Habib AG. Neutralization of cholera toxin with nanoparticle decoys for treatment of cholera. PLoS Neglected Trop Dis. 2018;12:e0006266. doi: 10.1371/journal.pntd.0006266.
- Kim EJ, Lee CH, Nair GB, Kim DW. Whole-genome sequence comparisons reveal the evolution of Vibrio cholerae O1. Trends Microbiol. 2015;23:479–489. doi: 10.1016/j.tim.2015.03.010.
- Johnson AM, Kaushik RS, Francis DH, Fleckenstein JM, Hardwidge PR. Heat-labile enterotoxin promotes escherichia coli adherence to intestinal epithelial cells. J Bacteriol. 2009;191:178–186.
- Duan Q, Pang S, Feng L, Liu J, Lv L, Li B, Liang Y, Zhu G. Heat-labile enterotoxin enhances F4-producing enterotoxigenic E. coli adhesion to porcine intestinal epithelial cells by upregulating bacterial adhesins and STb enterotoxin. Vet Res. 2022;53:88. doi: 10.1186/s13567-022-01110-4.
- Allen KP, Randolph MM, Fleckenstein JM. Importance of heat-labile enterotoxin in colonization of the adult mouse small intestine by human enterotoxigenic escherichia coli strains. Infect Immun. 2006;74:869–875. doi: 10.1128/IAI.74.2.869-875.2006.
- Das R, Nasrin S, Palit P, Sobi RA, Sultana A, Khan SH, Haque MA, Nuzhat S, Ahmed T, Faruque ASG, et al. Vibrio cholerae in rural and urban Bangladesh, findings from hospital-based surveillance, 2000–2021. NatSR. 2023;13:6411. doi: 10.1038/s41598-023-33576-3.
- Khalil I, Anderson JD, Bagamian KH, Baqar S, Giersing B, Hausdorff WP, Marshall C, Porter CK, Walker RI, Bourgeois AL. Vaccine value profile for enterotoxigenic Escherichia coli (ETEC). Vaccine. 2023;41:S95–S113. doi: 10.1016/j.vaccine.2023.02.011.
- López-Gigosos R, García-Fortea P, Reina-Doña E, Plaza-Martín E. Effectiveness in prevention of travellers’ diarrhoea by an oral cholera vaccine WC/rBS. Travel Med Infect Dis. 2007;5:380–384. doi: 10.1016/j.tmaid.2007.06.001.
- Clemens JD, Sack DA, Harris JR, Chakraborty J, Neogy PK, Stanton B, Huda N, Khan MU, Kay BA, Ansaruzzaman M, et al. Cross-protection by b subunit-whole cell cholera vaccine against diarrhea associated with heat-labile toxin-producing enterotoxigenic escherichia coli: results of a large-scale field trial. J Infect Dis. 1988;158:372–377. doi: 10.1093/infdis/158.2.372.
- Wierzba TF. Oral cholera vaccines and their impact on the global burden of disease. Hum Vaccines Immunother. 2018;15:1294–1301. doi: 10.1080/21645515.2018.1504155.
- Odevall L, Hong D, Digilio L, Sahastrabuddhe S, Mogasale V, Baik Y, Choi S, Kim JH, Lynch J. The Euvichol story – development and licensure of a safe, effective and affordable oral cholera vaccine through global public private partnerships. Vaccine. 2018;36:6606–6614. doi: 10.1016/j.vaccine.2018.09.026.
- Carlin N, Svennerholm A-M. Oral vaccines for enterotoxigenic Escherichia coli. In H Kiyono, D. W Pascual (Eds.), Mucosal Vaccines (Second Edition). 2020. pp. 563–578 Academic Press 10.1016/B978-0-12-811924-2.00032-8.
- Kantele A, Riekkinen M, Jokiranta TS, Pakkanen SH, Pietilä J, Patjas A, Eriksson M, Khawaja T, Klemets P, Marttinen K, et al. Safety and immunogenicity of ETVAX®, an oral inactivated vaccine against enterotoxigenic Escherichia coli diarrhoea: a double-blinded, randomized, placebo-controlled trial amongst Finnish travellers to Benin, West Africa. J Travel Med. 2023;30:taad045. doi: 10.1093/jtm/taad045.
- Taha AM, Mahmoud H, Hassan EM, Ghonaim MM. Harmonizing vaccine and infrastructure development to tame cholera outbreaks across Africa. Nat Commun. 2024;15:4686. doi: 10.1038/s41467-024-49077-4.
- Naylor C, Lu M, Haque R, Mondal D, Buonomo E, Nayak U, Mychaleckyj JC, Kirkpatrick B, Colgate R, Carmolli M, et al. Environmental enteropathy, oral vaccine failure and growth faltering in infants in Bangladesh. EBioMedicine. 2015;2:1759–1766. doi: 10.1016/j.ebiom.2015.09.036.
- Marie C, Ali A, Chandwe K, Petri WA, Kelly P. Pathophysiology of environmental enteric dysfunction and its impact on oral vaccine efficacy. Mucosal Immunol. 2018;11:1290–1298. doi: 10.1038/s41385-018-0036-1.
- Hayman B, Kumar Suri R, Downham M. Sustainable vaccine manufacturing in low- and middle-Income countries. Vaccine. 2022;40:7288–7304. doi: 10.1016/j.vaccine.2022.10.048.
- Venkatesan P. New measures to tackle the global cholera surge. Lancet Microbe. 2024;5:632. doi: 10.1016/S2666-5247(24)00139-9.
- Pukin AV, Branderhorst HM, Sisu C, Weijers CAGM, Gilbert M, Liskamp RMJ, Visser GM, Zuilhof H, Pieters RJ. Strong inhibition of cholera toxin by multivalent GM1 derivatives. Chembiochem Eur J Chem Biol. 2007;8:1500–1503. doi: 10.1002/cbic.200700266.
- Sisu C, Baron AJ, Branderhorst HM, Connell SD, Weijers CAGM, de Vries R, Hayes ED, Pukin AV, Gilbert M, Pieters RJ, et al. The influence of ligand valency on aggregation mechanisms for inhibiting bacterial toxins. Chembiochem. 2009;10:329–337. doi: 10.1002/cbic.200800541.
- Branson TR, Turnbull WB. Bacterial toxin inhibitors based on multivalent scaffolds. Chem Soc Rev. 2013;42:4613–4622. doi: 10.1039/c2cs35430f.
- Alsharif G, Ahmad S, Islam MS, Shah R, Busby SJ, Krachler AM. Host attachment and fluid shear are integrated into a mechanical signal regulating virulence in Escherichia coli O157:H7. Proc Natl Acad Sci U S A. 2015;112:5503–5508. doi: 10.1073/pnas.1422946112.
- Delon LC, Guo Z, Oszmiana A, Chien C, Gibson R, Prestidge C, Thierry B. A systematic investigation of the effect of the fluid shear stress on Caco-2 cells towards the optimization of epithelial organ-on-chip models. Biomaterials. 2019;225:119521. doi: 10.1016/j.biomaterials.2019.119521.
- Rivera-Chávez F, Mekalanos JJ. Cholera toxin promotes pathogen acquisition of host-derived nutrients. Nature (London). 2019;572:244–248. doi: 10.1038/s41586-019-1433-3.
- Holmgren J, Bourgeois L, Carlin N, Clements J, Gustafsson B, Lundgren A, Nygren E, Tobias J, Walker R, Svennerholm A. Development and preclinical evaluation of safety and immunogenicity of an oral ETEC vaccine containing inactivated E. coli bacteria overexpressing colonization factors CFA/I, CS3, CS5 and CS6 combined with a hybrid LT/CT B subunit antigen, administered alone and together with dmLT adjuvant. Vaccine. 2013;31:2457–2464. doi: 10.1016/j.vaccine.2013.03.027.
- Svennerholm A-M, Lundgren A, Leach S, Akhtar M, Qadri F. Mucosal immune responses against an oral enterotoxigenic escherichia coli vaccine evaluated in clinical trials. J Infect Dis. 2021;224:S821–S828. doi: 10.1093/infdis/jiab475.
- Tokuhara D, Yuki Y, Nochi T, Kodama T, Mejima M, Kurokawa S, Takahashi Y, Nanno M, Nakanishi U, Takaiwa F, et al. Secretory IgA-mediated protection against V. cholerae and heat-labile enterotoxin-producing enterotoxigenic Escherichia coli by rice-based vaccine. Proc Natl Acad Sci U S A. 2010;107:8794–8799. doi: 10.1073/pnas.0914121107.
- Hui Xian T, Parasuraman S, Ravichandran M, Prabhakaran G. Dual-use vaccine for diarrhoeal diseases: cross-protective immunogenicity of a cold-chain-free, live-attenuated, oral cholera vaccine against enterotoxigenic escherichia coli (ETEC) challenge in BALB/c mice. Vaccines. 2022;10:2161. doi: 10.3390/vaccines10122161.
- Lundgren A, Jertborn M, Svennerholm A-M. Induction of long term mucosal immunological memory in humans by an oral inactivated multivalent enterotoxigenic Escherichia coli vaccine. Vaccine. 2016;34:3132–3140. doi: 10.1016/j.vaccine.2016.04.055.
- Sarker SA, Jäkel M, Sultana S, Alam NH, Bardhan PK, Chisti MJ, Salam MA, Theis W, Hammarström L, Frenken LG. Anti-rotavirus protein reduces stool output in infants with diarrhea: a randomized placebo-controlled trial. Gastroenterology. 2013;145:740–748. doi: 10.1016/j.ebiom.2015.12.023.
- Steeland S, Vandenbroucke RE, Libert C. Nanobodies as therapeutics: big opportunities for small antibodies. Drug Discovery Today. 2016;21:1076–1113. doi: 10.1016/j.drudis.2016.04.003.
- Fernández-Quintero ML, Ljungars A, Waibl F, Greiff V, Andersen JT, Gjølberg TT, Jenkins TP, Voldborg BG, Grav LM, Kumar S, et al. Assessing developability early in the discovery process for novel biologics. Mabs. 2023;15:2171248. doi: 10.1080/19420862.2023.2171248.
- Validogen. Case study III - methanol-induced bivalent VHH production in Pichia yielding 31 g/L. 2024. https://www.validogen.com/news-events/detail/case-study-iii-methanol-induced-bivalent-vhh-production.
- Cherry JR, Fidantsef AL. Directed evolution of industrial enzymes: an update. Curr Opin Biotechnol. 2003;14:438–443. doi: 10.1016/S0958-1669(03)00099-5.
- Li C, Zhou J, Du G, Chen J, Takahashi S, Liu S. Developing Aspergillus niger as a cell factory for food enzyme production. Biotech Adv. 2020;44:107630. doi: 10.1016/j.biotechadv.2020.107630.
- Whaley KJ, Zeitlin L. Emerging antibody-based products for infectious diseases: planning for metric ton manufacturing. Hum Vaccines Immunother. 2022|2021;18:1930847. doi: 10.1080/21645515.2021.1930847.
- Joffré E, von Mentzer A, Abd El Ghany M, Oezguen N, Savidge T, Dougan G, Svennerholm A, Sjöling Å, Christie PJ. Allele variants of enterotoxigenic escherichia coli heat-labile toxin are globally transmitted and associated with colonization factors. J Bacteriol. 2014;197:392–403. doi: 10.1128/JCM.02050-14.
- Mudrak B, Kuehn MJ. Heat-labile enterotoxin: beyond GM1 binding. Toxins. 2010;2:1445–1470. doi: 10.3390/toxins2061445.
- Chakraborty S, Johura F, Sultana M, Zhang X, Sadique A, George C, Monira S, Sack D, Alam M. Epidemiology of enterotoxigenic escherichia coli among children and adults seeking care at hospitals in two geographically distinct rural areas in Bangladesh. Microorganisms. 2024;12:359. doi: 10.3390/microorganisms12020359.
- von Mentzer A, Connor TR, Wieler LH, Semmler T, Iguchi A, Thomson NR, Rasko DA, Joffre E, Corander J, Pickard D, et al. Identification of enterotoxigenic Escherichia coli (ETEC) clades with long-term global distribution. Nature Genet. 2014;46:1321–1326. doi: 10.1038/ng.3145.
- Smith HW. The exploitation of transmissible plasmids to study the pathogenesis of E. coli diarrhoea. Proc R Soc Med. 1973;66:272–273.
- Turner SM, Chaudhuri RR, Jiang Z, DuPont H, Gyles C, Penn CW, Pallen MJ, Henderson IR. Phylogenetic comparisons reveal multiple acquisitions of the toxin genes by enterotoxigenic escherichia coli strains of different evolutionary lineages. J Clin Microbiol. 2006;44:4528–4536. doi: 10.1128/JCM.01474-06.
- Fleckenstein JM, Sheikh A. Emerging themes in the molecular pathogenesis of enterotoxigenic escherichia coli. J Infect Dis. 2021;224:S813–S820. doi: 10.1093/infdis/jiab359.
- Wang H, Zhong Z, Luo Y, Cox E, Devriendt B. Heat-stable enterotoxins of enterotoxigenic escherichia coli and their impact on host immunity. Toxins. 2019;11:24. doi: 10.3390/toxins11010024.
- Haque MA, Nasrin S, Palit P, Das R, Wahid BZ, Gazi MA, Mahfuz M, Golam Faruque AS, Ahmed T. Site-specific analysis of the incidence rate of enterotoxigenic escherichia coli infection elucidates an association with childhood stunting, wasting, and being underweight: a secondary analysis of the MAL-ED birth cohort. Am J Trop Med Hyg. 2023;108:1192–1200. doi: 10.4269/ajtmh.22-659.
- Read LT, Hahn RW, Thompson CC, Bauer DL, Norton EB, Clements JD, Payne SM. Simultaneous exposure to escherichia coli heat-labile and heat-stable enterotoxins increases fluid secretion and alters cyclic nucleotide and cytokine production by intestinal epithelial Cells. Infect Immun. 2014;82:5308–5316. doi: 10.1128/IAI.02496-14.
- Bölin I, Bölin I, Wiklund G, Qadri F, Torres O, Bourgeois AL, Savarino S, Svennerholm A. Enterotoxigenic escherichia coli with STh and STp genotypes is associated with diarrhea both in children in areas of endemicity and in travelers. J Clin Microbiol. 2006;44:3872–3877. doi: 10.1128/JCM.00790-06.
- Kipkirui E, Koech M, Ombogo A, Kirera R, Ndonye J, Kipkemoi N, Kirui M, Philip C, Roth A, Flynn A, et al. Molecular characterization of enterotoxigenic Escherichia coli toxins and colonization factors in children under five years with acute diarrhea attending Kisii teaching and referral hospital, Kenya. Trop Dis Travel Med Vaccines. 2021;7:31. doi: 10.1186/s40794-021-00157-z.
- Turunen K, Antikainen J, Lääveri T, Kirveskari J, Svennerholm A, Kantele A. Clinical aspects of heat-labile and heat-stable toxin-producing enterotoxigenic Escherichia coli: a prospective study among Finnish travellers. Travel Med Infect Dis. 2020;38:101855. doi: 10.1016/j.tmaid.2020.101855.
- Holmner Å, Askarieh G, Ökvist M, Krengel U. Blood group antigen recognition by Escherichia coli heat-labile enterotoxin. J Mol Biol. 2007;371:754–764. doi: 10.1016/j.jmb.2007.05.064.
- Heim JB, Hodnik V, Heggelund JE, Anderluh G, Krengel U. Crystal structures of cholera toxin in complex with fucosylated receptors point to importance of secondary binding site. NatSR. 2019;9:12243. doi: 10.1038/s41598-019-48579-2.
- Heggelund JE, Heim JB, Bajc G, Hodnik V, Anderluh G, Krengel U. Specificity of Escherichia coli heat-labile enterotoxin investigated by single-site mutagenesis and crystallography. Int J Mol Sci. 2019;20:703. doi: 10.3390/ijms20030703.
- Cervin J, Wands AM, Casselbrant A, Wu H, Krishnamurthy S, Cvjetkovic A, Estelius J, Dedic B, Sethi A, Wallom K, et al. GM1 ganglioside-independent intoxication by Cholera toxin. PLoS Pathog. 2018;14:e1006862. doi: 10.1371/journal.ppat.1006862.
- Wands AM, Cervin J, Huang H, Zhang Y, Youn G, Brautigam CA, Matson Dzebo M, Björklund P, Wallenius V, Bright DK, et al. Fucosylated molecules competitively interfere with cholera toxin binding to host cells. ACS Infect Dis. 2018;4:758–770. doi: 10.1021/acsinfecdis.7b00085.
- Esmail S, Manolson MF. Advances in understanding N-glycosylation structure, function, and regulation in health and disease. EJCB. 2021;100:151186. doi: 10.1016/j.ejcb.2021.151186.
- Phipps KR, Patel S, Scaife K, Holmes T, Šoltésová A, Thrane SW, Vigsnæs LK, Baldwin N, Jørgensen C. Allergenicity, genotoxicity and subchronic toxicity assessment of IgG binding protein LT produced from Aspergillus oryzae. JAT. 2025;45:1614–1636. doi: 10.1002/jat.4787.
- Fekete PZ, Mateo KS, Zhang W, Moxley RA, Kaushik RS, Francis DH. Both enzymatic and non-enzymatic properties of heat-labile enterotoxin are responsible for LT-enhanced adherence of enterotoxigenic Escherichia coli to porcine IPEC-J2 cells. Vet Microbiol. 2013;164:330–335. doi: 10.1016/j.vetmic.2013.02.019.
- Qadri F, Saha A, Ahmed T, Al Tarique A, Begum YA, Svennerholm A. Disease burden due to enterotoxigenic Escherichia coli in the first 2 years of Life in an urban community in Bangladesh. Infect Immun. 2007;75:3961–3968. doi: 10.1128/IAI.00459-07.
- Holmner Å, Mackenzie A, Krengel U. Molecular basis of cholera blood-group dependence and implications for a world characterized by climate change. FEBS Lett. 2010;584:2548–2555. doi: 10.1016/j.febslet.2010.03.050.
- Laustsen AH, Greiff V, Karatt-Vellatt A, Muyldermans S, Jenkins TP. Animal immunization, in vitro display technologies, and machine learning for antibody discovery. Trends Biotechnol. 2021;39:1263–1273. doi: 10.1016/j.tibtech.2021.03.003.
- Wang Y, Huang Y, Chase RC, Li T, Ramai D, Antwi SO, Keaveny AP, Pang M. Global burden of digestive diseases: a systematic analysis of the global burden of diseases study, 1990 to 2019. Gastroenterology. 2023;165:773–783.e15. doi: 10.1053/j.gastro.2023.05.050.
Disclosure of Potential Conflicts of Interest
All authors affiliated with Bactolife A/S are present or past employees of Bactolife A/S. A.H.L. and S.W.T. are shareholders of Bactolife A/S.
Funding
We thank Innovation Fund Denmark (grant 1044-00106B) for their financial support to M.P. and J.S.P. (grant 1061-00001A), as well as the MICA Foundation for support to J.S.P. We also thank UiO for funding the position of N.M. SEC analysis, as well as the associated LT and CTX production, was carried out at the UiO Structural Biology core facilities, which are part of the Norwegian Macromolecular Crystallography Consortium (NORCRYST) and which received funding from the Norwegian INFRASTRUKTUR-program (project no. 245828) as well as from UiO (core facility funds).
Author contributions
M.P., S.W.T., A.H.L. and L.G. conceptualized the project. M.P. and J.S.P drafted the manuscript. T.E.A. and J.M.L. provided the flow chamber model and facilitated analysis. J.S.P. analyzed ETEC virulence in the flow chamber model. M.P. and H.B.H. determined blocking capacity of toxin-receptor interactions. A.D. and H.B.H. performed and analyzed binding affinities using SPR. A.B.W. facilitated in silico studies. M.L.F.-Q. and T.P.J. performed and analyzed the in silico epitope predictions. N. J. B. conducted affinity studies using BLI. N.M. produced and purified the holotoxins, performed size-exclusion chromatography analysis and contributed to the structural analysis, supervised by U. K. All authors were a part of the manuscript process and final review.
ORCID
Marcus Petersson 0000-0002-2466-9038
Jens Sivkær Pettersen 0000-0002-0293-6692
Ágnes Duzs 0000-0001-6176-3149
Monica L. Fernández-Quintero 0000-0002-6811-6283
Nick. J. Burlet 0009-0002-5099-3971
Ute Krengel 0000-0001-6688-8151
Timothy P. Jenkins 0000-0003-2979-5663
Andrew B. Ward 0000-0001-7153-3769
Thomas Emil Andersen 0000-0001-9453-1141
Jakob Møller-Jensen 0000-0002-6008-3771
Lone Gram 0000-0002-1076-5723
Andreas H. Laustsen 0000-0001-6918-5574
Sandra Wingaard Thrane 0000-0003-2098-2710



